Take a look at the Recent articles
Abstract
Purpose: To report a case of oculocutaneous hypopigmentation found in an infant with sever hypotonia, cardiomyopathy and agenesis of the corpus.
Case Report: Badanie to dotyczyło dziecka płci męskiej, które zostało urodzone po okresie ciąży 37 tygodni. Produkt zdrowych rodziców Yamani z opóźnieniem wzrostu, wrodzoną zaćmą obustronną, dysmorfią twarzy, jasnymi włosami i skórą, oczopląsem i miał przerost lewej komory serca. Mutacje w genie epg5 zostały zidentyfikowane jako przyczyna zespołu Vici.
Wniosek: Zespół Vici jest odrębną jednostką kliniczną. Jego główne objawy kliniczne to hipotonia, opóźnienie rozwoju, albinizm, zaćma, kardiomiopatia, niedobór odporności i agenezja ciała modzelowatego.
Słowa kluczowe
Zespół Vici; agenezja ciała modzelowatego; EPG5
Wprowadzenie
Zespół Vici jest ciężkim, recesywnie dziedziczonym zaburzeniem wrodzonym charakteryzującym się głównymi cechami agenezji ciała modzelowatego (ACC), zaćmą, hipopigmentacją skóry wokół oczu, kardiomiopatią, niepowodzeniem w rozwoju i zmienną.
Po raz pierwszy został opisany w 1988 roku, u dwóch braci z zespołem wad rozwojowych składającym się z agenezji ciała modzelowatego, skórnej hipopigmentacji, obustronnej zaćmy, rozszczepu wargi i podniebienia oraz złożonego niedoboru odporności. Rodzeństwo cierpiało na ciężkie opóźnienie psychoruchowe, drgawki, nawracające ciężkie infekcje dróg oddechowych oraz przewlekłą kandydozę śluzówkowo-skórną. Zmarli na bronchopneumonię w wieku 2 i 3 lat .
Zespół Vici jest zaburzeniem wielosystemowym związanym z wadliwą autofagią i sugeruje fundamentalną rolę szlaku autofagii w układzie odpornościowym oraz anatomicznym i funkcjonalnym tworzeniu narządów takich jak mózg i serce .
Raport kliniczny
To dziecko jest pierwszym produktem zdrowych spokrewnionych rodziców Yamani. Był pełnowartościowy, po bezproblemowej ciąży z normalnym porodem wierzchołkowym w 36 tygodniu ciąży, z masą urodzeniową 3,2 kg, obwodem czaszki 31 cm i punktacją Apgar 8/8.
W wieku 25 dni był hospitalizowany z historią kaszlu, duszności i gorączki przez ostatnie trzy dni. W połączeniu z sinicą, pacjent miał historię słabego ssania i nawracających ataków krztuszenia się przy każdym karmieniu od urodzenia, ale niestety był zaniedbywany. W wywiadzie brak wymiotów, wysypki skórnej i nieprawidłowych ruchów. Brak historii rodzinnej poronień lub zgonów.
W badaniu wyglądał blado, kachektycznie i dysmorficznie (Rycina 1). (hipopegmentacja skóry, oczu i włosów blond, szerokie czoło, hiperbolizm, długi philtrum i micrognathia). Jego waga wynosiła 4100 g (3-10 centyl), długość 53 cm (3-10 centyl), a obwód głowy 33 cm (3 centyl). W badaniu okulistycznym stwierdzono łagodny oczopląs poziomy, zaćmę obustronną oraz siatkówkę z flarą. Pełne krepitacje w klatce piersiowej obustronne, bez szmerów, z prawidłowym dźwiękiem serca i z głęboką hipotonią.
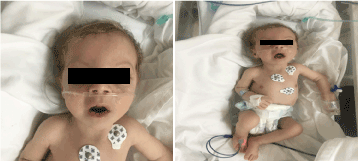
Rycina 1. Zdjęcie kliniczne pacjentki w wieku 2 miesięcy (a) hipotonia, (b) blond włosy, długi fartuch i mikrognacja
Badania laboratoryjne wykazały niedokrwistość mikrocytarną i hipochromiczną, profile nerkowe i krzepliwość były bez zmian. Transaminaza alaninowa (ALT) i aminotransferaza asparaginianowa (AST) były podwyższone odpowiednio 183-231 (10-45 U/L) i 193-301 (10-45 U/L). Poziom bilirubiny był prawidłowy. CK: 389 U/L i LDH: 612 U/L. Poziom amoniaku był prawidłowy i wynosił 13 mmol/l. W badaniu immunologicznym stwierdzono obniżony poziom immunoglobulin IgA: 0,1 g/L (0,7-4), ale IgG: 4,51 g/L (7-16), IgM: .8 g/L (0,4-2,3). mieściły się w granicach normy. Liczba limfocytów (CD3, CD4, CD8, CD19 i CD22) była prawidłowa. Krew, mocz i nie było wzrostu. Testy serologiczne na toksoplazmę, różyczkę, cytomegalię, herpes simplex i markery wątrobowe były w normie. W badaniu radiologicznym klatki piersiowej stwierdzono nacieki płucne oraz kardiomegalię. W badaniu ultrasonograficznym jamy brzusznej stwierdzono obustronne wodonercze. W badaniu echokardiograficznym stwierdzono przerost lewej komory serca. Rezonans magnetyczny (MRI) mózgowia potwierdził agenezję ciała modzelowatego (ryc. 2). Badania genetyczne potwierdziły homozygotyczny wariant utraty funkcji EPG5 (EPG5:NM_020964:exon9:c.1924C>T:p.R642X)
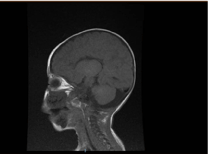
Rycina 2. MRI mózgu, przekrój strzałkowy ważony T1 pokazujący agenezję ciała modzelowatego
W 2. tygodniu od przyjęcia stan pacjenta szybko się pogarszał, z powtarzającymi się epizodami aspiracyjnego zapalenia płuc, które wymagały wentylacji. Ostatecznie u pacjenta doszło do zatrzymania krążenia i zgonu.
Materiały i metody
WES jest wykonywana na genomowym DNA przy użyciu przepływu pracy Agilent Sure Select Target Enrichment w celu wychwycenia regionów zainteresowania z biblioteki fragmentów DNA. Cały egzom jest sekwencjonowany na systemie sekwencjonowania Illumina HiSeq 2500 z minimalnym pokryciem 30X 95% regionów docelowych. Sekwencje egzomowego DNA probanta są mapowane i porównywane z sekwencją referencyjną genomu ludzkiego UCSC hg19. Saudi Diagnostic Laboratories wykorzystuje wewnętrzny potok do porównania sekwencji probanta z sekwencją referencyjną. Przeprowadzana jest ocena pokrycia i jakości dla wybranych kodujących eksonów znanych genów kodujących białka RefSeq. Analizy egzomów przesłuchują tysiące wariantów genetycznych u probanta przy użyciu zastrzeżonych baz danych dostosowanych do populacji arabskich. Ich podzbiory są charakteryzowane przy użyciu wytycznych American College of Medical Genetics and Genomics (ACMG) (Richards, et al. 2015) w celu sklasyfikowania ich znaczenia klinicznego. Sekwencjonowanie metodą Sangera wykonano w celu walidacji wszystkich wariantów uwzględnionych w raporcie, chyba że stwierdzono inaczej
Wyniki
U tego pacjenta zidentyfikowano homozygotyczny wariant utraty funkcji EPG5 (EPG5:NM_020964:exon9:c.1924C>T:p.R642X). Zgodnie z wytycznymi ACMG (Gen in Med 17:5, 2015) powinien on zostać zakwalifikowany jako wariant patogenny. Homozygotyczne warianty LOF (Loss of Function) EPG5 są częstym mechanizmem zespołu Vici, 242840 .
Dyskusja
Zespół Vici, jest rzadkim wrodzonym zaburzeniem wieloukładowym, które jest dziedziczone w sposób autosomalny recesywny, charakteryzuje się niepowodzeniem w rozwoju, hipopigmentacją, defektami immunologicznymi widocznymi przez nawracające infekcje, nieprawidłowościami neurologicznymi w postaci opóźnienia rozwoju, napadów drgawkowych, zaćmy, kardiomiopatii i nieprawidłowości nerwowo-mięśniowych; przy czym późniejsze zostały opisane tylko w kilku ostatnich opisach przypadków. Zespół Vici został po raz pierwszy opisany w 1988 roku przez Dionisi Vici, et al. w przypadku dwóch braci prezentujących albinizm, ACC, zaćmę, kardiomiopatię, opóźnienie umysłowe i niedobór odporności, a następnie został nazwany „zespołem Vici” przez del Campo, et al. w 1999 roku. Zespół Vici obserwowano zarówno u mężczyzn, jak i u kobiet, a w genie EPG5 zidentyfikowano szereg powiązanych mutacji
Nasi rodzice pacjentów są spokrewnieni i wspierają dziedziczenie autosomalne recesywne zespołu Vici. Trzynastu z 31 pacjentów, w tym nasz, to rodzeństwo. Opisano przypadki, w których rodzice byli dalekimi kuzynami .
Zespół Vici jest spowodowany recesywnymi mutacjami w EPG5 na chromosomie 18q12.3, kodującym ektopowe białko granulek P 5 (EPG5), kluczowy regulator autofagii w organizmach wyższych. Autofagia jest podstawową komórkową ścieżką degradacyjną zachowaną w toku ewolucji, odgrywającą ważną rolę w usuwaniu wadliwych białek i organelli, obronie przed infekcjami i adaptacji do zmieniających się wymagań metabolicznych .
Rozwój OUN, regulacja immunologiczna i pigmentacja skóry u pacjentów z zespołem Vici wskazują na udział autofagii w szerszym zakresie procesów komórkowych. W mózgu, autofagia została szeroko zbadana jako kluczowy mechanizm patogenetyczny w różnych zaburzeniach neurodegeneracyjnych .
Większość oznak i objawów zespołu vici jest obecna przy urodzeniu; niektóre cechy, takie jak zaćma, kardiomiopatia i niedobór odporności nie zawsze są obecne przy urodzeniu, ale oczekuje się, że będą ewoluować w ciągu pierwszych lat. A większość pacjentów zmarła w ciągu pierwszych trzech lat życia wtórnie do kardiomiopatii lub zapalenia płuc
Podziękowania
Autorzy pragną podziękować rodzicom pacjenta i laboratorium diagnostycznemu Saudi.
- Dionisi Vici C, Sabetta G, Gambarara M, Vigevano F, Bertini E, et al. (1988) Agenesis of the corpus callosum, combined immunodeficiency, bilateral cataract, and hypopigmentation in two brothers. Am J Med Genet 29:1-8.
- Thomas Cullup, Ay Lin Kho, Carlo Dionisi-Vici (2013) Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nature Genetics 45: 83-87.
- Said E, Soler D, Sewry C (2012) Vici syndrome-a rapidly progressive neurodegenerative disorder with hypopigmentation, immunodeficiency and myopathic changes on muscle biopsy. Am J Med Genet A 158: 440-444.
- del Campo M, Hall BD, Aeby A, Nassogne MC, Verloes A, et al. (1999) Albinizm i agenezja ciała modzelowatego z głębokim opóźnieniem rozwoju: Vici syndrome, evidence for autosomal recessive inheritance. Am J Med Genet 85: 479-485.
- Ehmke N, Parvaneh N, Krawitz P, Ashrafi MR, Karimi P, et al. (2014) First description of a patient with Vici syndrome due to a mutation affecting the penultimate exon of EPG5 and review of the literature. Am J Med Genet A 164A: 3170-3175.
- Al-Owain M, Al-Hashem A, Al-Muhaizea M, Humaidan H, Al-Hindi H, et al. (2010) Vici syndrome associated with unilateral lung hypoplasia and myopathy. Am J Med Genet A 152A: 1849-1853.
- Byrne S, Dionisi-Vici C, Smith L, Gautel M, Jungbluth H, et al. (2016) Vici syndrome: a review. Orphanet J Rare Dis 11: 21.
- Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, et al. (2006) Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol 76: 89-101.
- Cullup T, Dionisi-Vici C, Kho AL, Yau S, Mohammed S, et al. (2014) Clinical utility gene card for: Vici Syndrome. Eur J Hum Genet 22.
- Curtis Rogers R, Bridgette Aufmuth, Stephanie Monesson (2011) Vici Syndrome: A Rare Autosomal Recessive Syndrome with Brain Anomalies, Cardiomyopathy, and Severe Intellectual Disability. Case Reports in Genetics 2011: 421582.