Dê uma olhada nos artigos recentes
Abstract
Propósito: Relatar um caso de hipopigmentação oculocutânea encontrado em um bebê com hipotonia grave, cardiomiopatia e agenesia do corpus.
>Abstract
Case Report: Este estudo envolveu um bebê do sexo masculino que deu à luz após um período gestacional de 37 semanas. Produto de pais Yamani consanguíneos saudáveis com retardo de crescimento, cataratas bilaterais congênitas e dismórficas faciais, cabelos e pele justos, nistagmo e hipertrofia ventricular esquerda. Mutações no gene epg5 foram identificadas como a causa da síndrome de Vici.
Conclusão: A síndrome de Vici é uma entidade clínica distinta. Suas principais manifestações clínicas incluem hipotonai, atraso no desenvolvimento, albinismo, catarata, cardiomiopatia, imunodeficiência e agenesia do corpo caloso.
Palavras-chave
Síndrome de Vici; agenesia do corpus callosum; EPG5
Introdução
Síndrome de Vici é uma doença congênita grave, recessivamente herdada, caracterizada pelas principais características de agenesia do corpus callosum (ACC), cataratas, hipopigmentação oculocutânea, cardiomiopatia, insucesso de desenvolvimento e variável.
Foi descrita pela primeira vez em 1988, em dois irmãos com síndrome de malformação que consiste na agenesia do corpo caloso, hipopigmentação cutânea, catarata bilateral, lábio leporino e palato fendido e imunodeficiência combinada. Os irmãos sofriam de retardo psicomotor grave, convulsões, infecções respiratórias graves recorrentes, e candidíase mucocutânea crônica. Morreram de broncopneumonia aos 2 e 3 anos de idade .
Síndrome de Vici é uma desordem multissistémica associada a uma autofagia defeituosa e sugere um papel fundamental da via da autofagia no sistema imunitário e na formação anatómica e funcional de órgãos como o cérebro e o coração .
>
Relatório Clínico
Este bebé é o primeiro produto de Yamani Parents saudáveis e consanguíneos. Ele estava em plena gestação, após uma gravidez sem intercorrências com parto normal aos 36 semanas de idade gestacional com peso ao nascer de 3,2 kg, circunferência craniana de 31 cm e pontuação de Apgar de 8/8,
Aos 25 dias de idade, ele foi hospitalizado com história de tosse, falta de ar e febre nos últimos três dias. Associado à cianose, o paciente tinha histórico de maus ataques de sucção e asfixia recorrente a cada alimentação desde o nascimento, mas infelizmente foi negligenciado. Sem histórico de vômitos ou erupção cutânea e sem histórico de movimentos anormais. Nenhum histórico familiar de abortos ou mortes.
No exame ele parecia pálido, caquético e dismórfico (Figura 1). (pele hipopegmentada, olhos e cabelos loiros, testa larga, hiperbolismo, philtrum longo e micrognatia). Seu peso era de 4.100 g (3-10º centilo), comprimento 53 cm (3-10º centilo), e circunferência da cabeça 33 cm (3º centilo). Nos exames de oftalmologia, os exames mostraram nistagmo horizontal leve, catarata bilateral e retina flair. Cheio de crepitação no peito de bilaterl, sem murmúrio com som cardíaco normal e com hipotonia profunda.
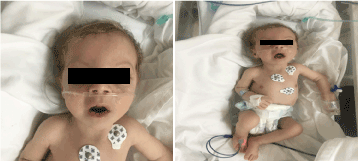
Figure 1. Foto clínica de paciente com 2 meses de idade (a) Hipotonia, (b) loiro de filtrum longo e micrognatia
>
A investigação laboratorial revelou anemia microcítica e hipocrómica, perfis renais e coagulação não notáveis. Alanina transaminase (ALT) e aspartato aminotransferase (AST) foram elevadas em 183-231 (10-45 U/L), e 193-301 (10-45 U/L), respectivamente. A bilirrubina estava normal. CK: 389 U/L e LDH: 612 U/L. O amoníaco era normal 13 mmol/L. O trabalho imunológico revelou níveis reduzidos de imunoglobulinas IgA: 0,1 g/L (0,7-4), mas a IgG: 4,51 g/L (7-16), IgM: .8 g/L (0,4-2,3). Estavam dentro do valor normal. As contagens de linfócitos (CD3, CD4, CD8, CD19, e CD22) estavam normais. Sangue, urina e não apresentaram crescimento. O teste serológico para toxoplasma, rubéola, citomegalovírus, herpes simples e marcadores hepáticos foram todos normais. A radiografia de tórax mostrou infiltração pneumónica e cardiomegalia. A ultra-sonografia abdominal revelou hidronefrose bilateral. A hipertrofia ventricular esquerda foi detectada pelo estudo ecocardiográfico. A ressonância magnética (RM) do cérebro confirmou agenesia do corpus calosum (Figura 2). Testes genéticos foram confirmados Uma perda homozigótica da variante funcional do EPG5 (EPG5:NM_020964:exon9:c.1924C>T:p.R642X)
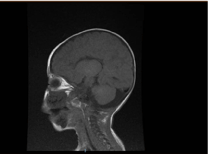
Figure 2. RM do cérebro, Sagittal T1 secção ponderada mostrando agenesia do corpo caloso
Durante a 2ª semana de admissão, a sua condição deteriorou-se rapidamente com repetidos episódios de pneumonia aspirativa que necessitaram de ventilação. Eventualmente, o paciente entrou em parada cardiopulmonar e morreu.
Materiais e métodos
OES é realizado em DNA genômico usando o fluxo de trabalho Agilent Sure Select Target Enrichment para capturar regiões de interesse a partir de uma biblioteca de fragmentos de DNA. Todo o exome é sequenciado no sistema sequenciador Illumina HiSeq 2500 com uma cobertura mínima de 30X de 95% das regiões alvo. As sequências de DNA do exoma da sonda são mapeadas e comparadas com a sequência de referência UCSC hg19 da construção do genoma humano. Os Laboratórios Saudi Diagnostic utilizam um pipeline interno para comparar a sequência da sonda com a sequência de referência. É feita uma avaliação da cobertura e da qualidade dos exons de codificação dos genes RefSeq de codificação de proteínas conhecidos. As análises exome interrogam milhares de variantes genéticas em uma probanda usando bases de dados proprietárias personalizadas para as populações árabes. Subconjuntos destes são caracterizados usando as diretrizes do American College of Medical Genetics and Genomics (ACMG) (Richards, et al. 2015) para classificar a sua relevância clínica. O sequenciamento Sanger é realizado para validar todas as variantes incluídas no relatório, a menos que seja indicado o contrário
Resultados
Perda homozigota da variante funcional do EPG5 (EPG5:NM_020964:exon9:c.1924C>T:p.R642X) foi identificada neste paciente. De acordo com as diretrizes do ACMG (Gen em Med 17:5, 2015) deve ser classificado como uma variante patogênica. As variantes homozigotos de perda de função (LOF) do EPG5 são um mecanismo comum da síndrome de Vici, 242840 .
Discussão
Síndrome Vici, é um distúrbio congênito raro do multissistema que é herdado em um padrão autossômico recessivo, é caracterizado por falha no desenvolvimento, hipopigmentação, defeitos imunológicos evidentes por infecções recorrentes, anormalidades neurológicas de atraso no desenvolvimento, convulsões, cataratas, cardiomiopatia e anormalidades neuromusculares; sendo as últimas descritas apenas em alguns relatos de casos recentes. A síndrome de Vici foi primeiramente relatada em 1988 por Dionisi Vici, et al. no caso de dois irmãos apresentando albinismo, ACC, cataratas, cardiomiopatia, retardo mental e imunodeficiência, e foi posteriormente denominada “síndrome de Vici” por del Campo, et al. em 1999. A síndrome Vici tem sido observada tanto em homens quanto em mulheres, e várias mutações relacionadas foram identificadas no gene EPG5 .
Nossos pais pacientes têm consanguinidade e sustentam herança autossômica recessiva da síndrome Vici. Treze dos 31 pacientes, incluindo os nossos, são irmãos. Foram relatados casos em que os pais eram primos distantes .
Síndrome de Vici é devido a mutações recessivas no EPG5 no cromossomo 18q12.3, codificando a proteína ectópica P granulado 5 (EPG5), um regulador chave de autofagia em organismos superiores. A autofagia é uma via degradativa celular fundamental conservada ao longo da evolução com papéis importantes na remoção de proteínas e organelas defeituosas, defesa contra infecções e adaptação a mudanças nas demandas metabólicas .
Desenvolvimento do SNS, regulação imunológica e pigmentação da pele em pacientes com síndrome de Vici implicam a autofagia em uma gama mais ampla de processos celulares. No cérebro, a autofagia tem sido amplamente investigada como um mecanismo patogênico chave em vários distúrbios neurodegenerativos .
Os sinais e sintomas da síndrome vici estão presentes ao nascimento; algumas características como catarata, cardiomiopatia e imunodeficiência nem sempre estão presentes ao nascimento, mas espera-se que evoluam ao longo dos primeiros anos . E a maioria dos pacientes morreu nos primeiros três anos de vida secundária à cardiomiopatia ou pneumonia
>
Agradecimentos
Os autores desejam agradecer aos pais do paciente e ao laboratório de diagnóstico saudita.
- Dionisi Vici C, Sabetta G, Gambarara M, Vigevano F, Bertini E, et al. (1988) Agenesia do corpus callosum, imunodeficiência combinada, catarata bilateral e hipopigmentação em dois irmãos. Am J Med Genet 29:1-8.
- Thomas Cullup, Ay Lin Kho, Carlo Dionisi-Vici (2013) Mutações recessivas no EPG5 causam síndrome de Vici, um distúrbio multissistêmico com autofagia defeituosa. Nature Genetics 45: 83-87.
- Said E, Soler D, Sewry C (2012) Síndrome de Vici – um distúrbio neurodegenerativo rapidamente progressivo com hipopigmentação, imunodeficiência e alterações miopáticas na biópsia muscular. Am J Med Genet A 158: 440-444.
- del Campo M, Hall BD, Aeby A, Nassogne MC, Verloes A, et al. (1999) Albinismo e agenesia do corpo caloso com profundo atraso de desenvolvimento: Síndrome de Vici, evidência de herança autossômica recessiva. Am J Med Genet 85: 479-485.
- Ehmke N, Parvaneh N, Krawitz P, Ashrafi MR, Karimi P, et al. (2014) Primeira descrição de um paciente com síndrome de Vici devido a uma mutação que afeta o penúltimo exon do EPG5 e revisão da literatura. Am J Med Genet A 164A: 3170-3175.
- Al-Owain M, Al-Hashem A, Al-Muhaizea M, Humaidan H, Al-Hindi H, et al. (2010) Síndrome de Vici associada a hipoplasia pulmonar unilateral e miopatia. Am J Med Genet A 152A: 1849-1853.
- Byrne S, Dionisi-Vici C, Smith L, Gautel M, Jungbluth H, et al. (2016) Síndrome de Vici: uma revisão. Orphanet J Rare Dis 11: 21.
- Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, et al. (2006) Aggregate-prone proteins are clear from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol 76: 89-101.
- Cullup T, Dionisi-Vici C, Kho AL, Yau S, Mohammed S, et al. (2014) Clinical utility gene card for: Síndrome de Vici. Eur J Hum Genet 22.
- Curtis Rogers R, Bridgette Aufmuth, Stephanie Monesson (2011) Síndrome de Vici: A Rare Autosomal Recessive Syndrome with Brain Anomalies, Cardiomyopathy, and Severe Intellectual Disability. Relatos de casos em Genética 2011: 421582.