Guarda gli articoli recenti
Abstract
Scopo: Riportare un caso di ipopigmentazione oculocutanea trovata in un neonato con grave ipotonia, cardiomiopatia e agenesia del corpo: Questo studio ha coinvolto un bambino maschio che è stato consegnato dopo un periodo gestazionale di 37 settimane. Prodotto da genitori Yamani consanguinei sani con ritardo di crescita, cataratta bilaterale congenita e dismorfismo del viso, capelli chiari e pelle, nistagmo e aveva ipertrofia del ventricolo sinistro. Mutazioni nel gene epg5 sono state identificate come causa della sindrome di Vici.
Conclusione: La sindrome di Vici è un’entità clinica distinta. Le sue principali manifestazioni cliniche includono ipotonia, ritardo nello sviluppo, albinismo, cataratta, cardiomiopatia, immunodeficienza e agenesia del corpo calloso.
parole chiave
Sindrome di Vici; agenesia del corpo calloso; EPG5
Introduzione
La sindrome di Vici è un grave disordine congenito ereditario recessivo caratterizzato dalle principali caratteristiche dell’agenesia del corpo calloso (ACC), cataratta, ipopigmentazione oculocutanea, cardiomiopatia, mancata crescita e variabile.
È stata descritta per la prima volta nel 1988, in due fratelli con una sindrome malformativa composta da agenesia del corpo calloso, ipopigmentazione cutanea, cataratta bilaterale, labiopalatoschisi e immunodeficienza combinata. I fratelli soffrivano di grave ritardo psicomotorio, convulsioni, gravi infezioni respiratorie ricorrenti e candidosi mucocutanea cronica. Sono morti di broncopolmonite all’età di 2 e 3 anni.
La sindrome di Vici è un disordine multisistemico associato all’autofagia difettosa e suggerisce un ruolo fondamentale della via dell’autofagia nel sistema immunitario e nella formazione anatomica e funzionale di organi come il cervello e il cuore.
Relazione clinica
Questo bambino è il primo prodotto di genitori consanguinei Yamani sani. Era a termine, dopo una gravidanza non movimentata con parto normale al vertice a 36 settimane di età gestazionale con peso alla nascita di 3,2 kg, una circonferenza cranica di 31 cm e un punteggio Apgar di 8/8.
A 25 giorni, è stato ricoverato con storia di tosse, mancanza di respiro e febbre per gli ultimi tre giorni. Associato a cianosi, il paziente aveva una storia di scarsa suzione e attacchi ricorrenti di soffocamento con ogni alimentazione dalla nascita, ma purtroppo è stato trascurato. Nessuna storia di vomito o eruzione cutanea e nessuna storia di movimenti anomali. Nessuna storia familiare di aborti o morti.
Nell’esame aveva un aspetto pallido, cachettico e dismorfo (Figura 1). (pelle ipopegmentata, occhi e capelli biondi, fronte larga, iperbolismo, filastrocca lunga e micrognazia). Il suo peso era di 4.100 g (3-10° centile), la lunghezza 53 cm (3-10° centile) e la circonferenza della testa 33 cm (3° centile). Gli esami oftalmologici hanno mostrato un lieve nistagmo orizzontale, cataratta bilaterale e retina a scaglie. Pieno di crepitazione sul petto bilaterale, nessun soffio con suono cardiaco normale e aveva profonda ipotonia.
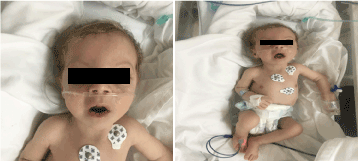
Figura 1. Foto clinica del paziente a 2 mesi di età (a) ipotonia, (b) biondo di capelli lungo filetto e micrognazia
L’indagine di laboratorio ha rivelato anemia microcitica e ipocromica, profili renali e coagulazione erano irrilevanti. L’alanina transaminasi (ALT) e l’aspartato aminotransferasi (AST) erano elevate rispettivamente a 183-231 (10-45 U/L) e 193-301 (10-45 U/L). La bilirubina era normale. CK: 389 U/L e LDH: 612 U/L. L’ammoniaca era normale 13 mmol/L. Il workup immunologico ha rivelato livelli ridotti di immunoglobuline IgA: 0,1 g/L (0,7-4), ma le IgG: 4,51 g/L (7-16), IgM: .8 g/L (0,4-2,3). Erano entro il valore normale. La conta dei sottoinsiemi di linfociti (CD3, CD4, CD8, CD19 e CD22) era normale. Sangue, urine e non erano in crescita. Il test sierologico per toxoplasma, rosolia, citomegalovirus, herpes simplex, e i marcatori epatici erano tutti normali. La radiografia del torace ha mostrato infiltrazione pneumonica e cardiomegalia. L’ecografia addominale ha rivelato un’idronefrosi bilaterale. L’ipertrofia ventricolare sinistra è stata rilevata dallo studio ecocardiografico. La risonanza magnetica (MRI) del cervello ha confermato l’agenesia del corpo calloso (Figura 2). Il test genetico è stato confermato Una variante omozigote di perdita di funzione di EPG5 (EPG5:NM_020964:esone9:c.1924C>T:p.R642X)
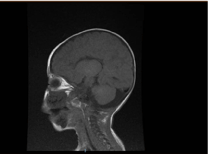
Figura 2. Risonanza magnetica del cervello, sezione sagittale T1 pesata che mostra l’agenesia del corpo calloso
Durante la seconda settimana di ricovero, le sue condizioni sono peggiorate rapidamente con ripetuti episodi di polmonite da aspirazione che hanno richiesto la ventilazione. Alla fine, il paziente è andato in arresto cardiopolmonare ed è morto.
Materiali e metodi
WES viene eseguito su DNA genomico utilizzando il flusso di lavoro Agilent Sure Select Target Enrichment per catturare regioni di interesse da una libreria di frammenti di DNA. L’intero esoma è sequenziato sul sistema di sequenziamento Illumina HiSeq 2500 con una copertura minima di 30X del 95% delle regioni target. Le sequenze di DNA dell’esoma del probando sono mappate e confrontate con la sequenza di riferimento UCSC hg19 del genoma umano. Saudi Diagnostic Laboratories utilizza una pipeline interna per confrontare la sequenza del probando con la sequenza di riferimento. Viene eseguita la valutazione della copertura e della qualità per gli esoni codificanti mirati dei geni RefSeq noti che codificano le proteine. Le analisi dell’esoma interrogano migliaia di varianti genetiche in un probando utilizzando database proprietari personalizzati per le popolazioni arabe. Sottoinsiemi di queste sono caratterizzati utilizzando le linee guida dell’American College of Medical Genetics and Genomics (ACMG) (Richards, et al. 2015) per classificare la loro rilevanza clinica. Il sequenziamento Sanger viene eseguito per convalidare tutte le varianti incluse nel rapporto se non diversamente specificato
Risultati
In questo paziente è stata identificata una variante omozigote di perdita di funzione di EPG5 (EPG5:NM_020964:esone9:c.1924C>T:p.R642X). Secondo le linee guida ACMG (Gen in Med 17:5, 2015) dovrebbe essere classificata come variante patogena. Le varianti omozigoti di perdita di funzione (LOF) di EPG5 sono un meccanismo comune della sindrome di Vici, 242840 .
Discussione
La sindrome di Vici, è un raro disordine congenito multi-sistema che è ereditato in un modello autosomico recessivo, è caratterizzato da mancata crescita, ipopigmentazione, difetti immunologici evidenti da infezioni ricorrenti, anomalie neurologiche di ritardo nello sviluppo, convulsioni, cataratta, cardiomiopatia e anomalie neuromuscolari; con la successiva solo essere descritto in pochi casi recenti. La sindrome di Vici è stata segnalata per la prima volta nel 1988 da Dionisi Vici, et al. nel caso di due fratelli che presentavano albinismo, ACC, cataratta, cardiomiopatia, ritardo mentale e immunodeficienza, ed è stata successivamente chiamata ‘sindrome di Vici’ da del Campo, et al. nel 1999. La sindrome di Vici è stata osservata sia nei maschi che nelle femmine, e una serie di mutazioni correlate sono state identificate nel gene EPG5.
I nostri genitori pazienti hanno consanguineità e sostengono l’eredità autosomica recessiva della sindrome di Vici. Tredici dei 31 pazienti, compreso il nostro, sono fratelli e sorelle. Sono stati riportati casi in cui i genitori erano lontani cugini.
La sindrome di Vici è dovuta a mutazioni recessive in EPG5 sul cromosoma 18q12.3, che codifica la proteina 5 dei granuli P ectopici (EPG5), un regolatore di autofagia chiave negli organismi superiori. L’autofagia è una fondamentale via degradativa cellulare conservata nel corso dell’evoluzione con ruoli importanti nella rimozione di proteine e organelli difettosi, nella difesa contro le infezioni e nell’adattamento alle mutevoli esigenze metaboliche.
Lo sviluppo del SNC, la regolazione immunitaria e la pigmentazione della pelle nei pazienti con la sindrome di Vici implicano l’autofagia in una gamma più ampia di processi cellulari. Nel cervello, l’autofagia è stata ampiamente studiata come un meccanismo patogenetico chiave in vari disturbi neurodegenerativi .
La maggior parte dei segni e dei sintomi della sindrome di Vici sono presenti alla nascita; alcune caratteristiche come la cataratta, la cardiomiopatia e l’immunodeficienza non sono sempre presenti alla nascita, ma si prevede che evolvano nel corso dei primi anni . E la maggior parte dei pazienti sono morti entro i primi tre anni di vita a causa di cardiomiopatia o polmonite
Riconoscimenti
Gli autori desiderano ringraziare i genitori del paziente e il laboratorio diagnostico Saudi.
- Dionisi Vici C, Sabetta G, Gambarara M, Vigevano F, Bertini E, et al. (1988) Agenesia del corpo calloso, immunodeficienza combinata, cataratta bilaterale e ipopigmentazione in due fratelli. Am J Med Genet 29:1-8.
- Thomas Cullup, Ay Lin Kho, Carlo Dionisi-Vici (2013) Mutazioni recessive in EPG5 causano la sindrome di Vici, un disturbo multisistemico con autofagia difettosa. Natura Genetica 45: 83-87.
- Said E, Soler D, Sewry C (2012) Vici syndrome-un disordine neurodegenerativo rapidamente progressivo con ipopigmentazione, immunodeficienza e cambiamenti miopatici sulla biopsia muscolare. Am J Med Genet A 158: 440-444.
- del Campo M, Hall BD, Aeby A, Nassogne MC, Verloes A, et al. (1999) Albinismo e agenesia del corpo calloso con profondo ritardo nello sviluppo: Sindrome di Vici, prova di eredità autosomica recessiva. Am J Med Genet 85: 479-485.
- Ehmke N, Parvaneh N, Krawitz P, Ashrafi MR, Karimi P, et al. (2014) Prima descrizione di un paziente con sindrome di Vici a causa di una mutazione che colpisce il penultimo esone di EPG5 e revisione della letteratura. Am J Med Genet A 164A: 3170-3175.
- Al-Owain M, Al-Hashem A, Al-Muhaizea M, Humaidan H, Al-Hindi H, et al. (2010) Vici sindrome associata a ipoplasia polmonare unilaterale e miopatia. Am J Med Genet A 152A: 1849-1853.
- Byrne S, Dionisi-Vici C, Smith L, Gautel M, Jungbluth H, et al. (2016) Vici syndrome: una revisione. Orphanet J Rare Dis 11: 21.
- Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, et al. (2006) Le proteine soggette ad aggregazione vengono eliminate dal citosol dall’autofagia: implicazioni terapeutiche. Curr Top Dev Biol 76: 89-101.
- Cullup T, Dionisi-Vici C, Kho AL, Yau S, Mohammed S, et al. (2014) Scheda gene utilità clinica per: Sindrome di Vici. Eur J Hum Genet 22.
- Curtis Rogers R, Bridgette Aufmuth, Stephanie Monesson (2011) Vici Syndrome: Una rara sindrome autosomica recessiva con anomalie cerebrali, cardiomiopatia e grave disabilità intellettuale. Case Reports in Genetics 2011: 421582.