Echa un vistazo a los artículos recientes
Abstract
Propósito: Informar de un caso de hipopigmentación oculocutánea encontrado en un lactante con hipotonía severa, cardiomiopatía y agenesia del corpus.
Informe de caso: Se trata de un bebé varón que nació después de un período de gestación de 37 semanas. Producto de padres Yamani consanguíneos sanos con retraso en el crecimiento, cataratas bilaterales congénitas, y facial dismórfico, pelo y piel claros, nistagmo y tenía hipertrofia del ventrículo izquierdo. Se han identificado mutaciones en el gen epg5 como causa del síndrome de Vici.
Conclusión: El síndrome de Vici es una entidad clínica distinta. Sus principales manifestaciones clínicas incluyen hipotonai, retraso en el desarrollo, albinismo, cataratas, cardiomiopatía, inmunodeficiencia y agenesia del cuerpo calloso.
Palabras clave
Síndrome de Vici; agenesia del cuerpo calloso; EPG5
Introducción
El síndrome de Vici es un trastorno congénito grave, de herencia recesiva, caracterizado por los rasgos principales de agenesia del cuerpo calloso (ACC), cataratas, hipopigmentación oculocutánea, cardiomiopatía, retraso en el crecimiento y variable.
Se describió por primera vez en 1988, en dos hermanos con un síndrome de malformación consistente en agenesia del cuerpo calloso, hipopigmentación cutánea, catarata bilateral, labio y paladar hendido e inmunodeficiencia combinada. Los hermanos padecían retraso psicomotor grave, convulsiones, infecciones respiratorias graves recurrentes y candidiasis mucocutánea crónica. Murieron de bronconeumonía a los 2 y 3 años de edad.
El síndrome de Vici es un trastorno multisistémico asociado a una autofagia defectuosa y sugiere un papel fundamental de la vía de la autofagia en el sistema inmunitario y en la formación anatómica y funcional de órganos como el cerebro y el corazón.
Informe clínico
Este bebé es el primer producto de padres Yamani consanguíneos sanos. Llegó a término, tras un embarazo sin incidentes con un parto de vértice normal a las 36 semanas de edad gestacional, con un peso al nacer de 3,2 kg, un perímetro craneal de 31 cm y una puntuación de Apgar de 8/8.
A los 25 días de vida, fue hospitalizado con historia de tos, dificultad respiratoria y fiebre durante los últimos tres días. Asociado a cianosis, el paciente tenía historia de mala succión y ataques de asfixia recurrentes con cada alimentación desde el nacimiento, pero desafortunadamente fue descuidado. No hay antecedentes de vómitos ni de erupciones cutáneas ni de movimientos anormales. No hay antecedentes familiares de abortos o muertes.
En la exploración tenía un aspecto pálido, caquéctico y dismórfico (Figura 1). (piel hipopegmentada, ojo y pelo rubio, frente ancha, hiperbolismo, filtrum largo y micrognatia). Su peso era de 4.100 g (percentil 3-10), su longitud de 53 cm (percentil 3-10) y su perímetro cefálico de 33 cm (percentil 3). En las exploraciones oftalmológicas se observaba un nistagmo horizontal leve, cataratas bilaterales y retina en forma de escamas. Lleno de crepitación en el tórax bilateral, sin soplo con sonido cardíaco normal y tenía una hipotonía profunda.
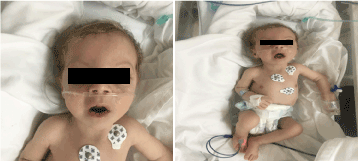
Figura 1. Foto clínica de la paciente a los 2 meses de edad (a) Hipotonía, (b) rubia de pelo largo filtrum y micrognatia
La investigación de laboratorio reveló anemia microcítica e hipocrómica, los perfiles renales y la coagulación no fueron notables. La alanina transaminasa (ALT) y la aspartato aminotransferasa (AST) estaban elevadas a 183-231 (10-45 U/L), y 193-301 (10-45 U/L), respectivamente. La bilirrubina era normal. CK: 389 U/L y LDH: 612 U/L. El amoníaco era normal 13 mmol/L. El estudio inmunológico reveló niveles reducidos de inmunoglobulinas IgA: 0,1 g/L (0,7-4), pero la IgG: 4,51 g/L (7-16), IgM: 0,8 g/L (0,4-2,3). Estaban dentro del valor normal. Los recuentos de subconjuntos de linfocitos (CD3, CD4, CD8, CD19 y CD22) eran normales. En la sangre, la orina y no había crecimiento. Las pruebas serológicas de toxoplasma, rubéola, citomegalovirus, herpes simple y marcadores hepáticos fueron normales. La radiografía de tórax mostró infiltración neumónica y cardiomegalia. La ecografía abdominal reveló una hidronefrosis bilateral. Se detectó hipertrofia ventricular izquierda mediante estudio ecocardiográfico. La resonancia magnética cerebral confirmó la agenesia del cuerpo calloso (figura 2). Las pruebas genéticas confirmaron una variante homocigótica de pérdida de función de EPG5 (EPG5:NM_020964:exon9:c.1924C>T:p.R642X)
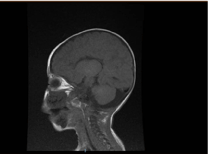
Figura 2. Resonancia magnética del cerebro, sección sagital ponderada en T1 que muestra agenesia del cuerpo calloso
Durante la segunda semana de ingreso, su estado se deterioró rápidamente con repetidos episodios de neumonía por aspiración que requirieron ventilación. Finalmente, el paciente entró en parada cardiopulmonar y falleció.
Materiales y métodos
El WES se realiza en ADN genómico utilizando el flujo de trabajo Agilent Sure Select Target Enrichment para capturar regiones de interés de una biblioteca de fragmentos de ADN. El exoma completo se secuencia en el sistema de secuenciación Illumina HiSeq 2500 con una cobertura mínima de 30X del 95% de las regiones objetivo. Las secuencias de ADN del exoma del probando se mapean y se comparan con la secuencia de referencia UCSC hg19 del genoma humano. Saudi Diagnostic Laboratories utiliza un proceso interno para comparar la secuencia del probando con la secuencia de referencia. Se realiza una evaluación de la cobertura y la calidad de los exones codificantes de los genes RefSeq conocidos que codifican proteínas. Los análisis del exoma interrogan a miles de variantes genéticas en un probando utilizando bases de datos propias adaptadas a las poblaciones árabes. Los subconjuntos de éstas se caracterizan utilizando las directrices del American College of Medical Genetics and Genomics (ACMG) (Richards, et al. 2015) para clasificar su relevancia clínica. Se realiza la secuenciación de Sanger para validar todas las variantes incluidas en el informe, a menos que se indique lo contrario
Resultados
En este paciente se identificó una variante homocigótica de pérdida de función de EPG5 (EPG5:NM_020964:exón9:c.1924C>T:p.R642X). Según las directrices del ACMG (Gen in Med 17:5, 2015) debe clasificarse como variante patogénica. Las variantes homocigóticas de pérdida de función (LOF) de EPG5 son un mecanismo común del síndrome de Vici, 242840 .
Discusión
El síndrome de Vici, es un raro trastorno multisistémico congénito que se hereda con un patrón autosómico recesivo, se caracteriza por retraso en el desarrollo, hipopigmentación, defectos inmunológicos evidentes por infecciones recurrentes, anomalías neurológicas de retraso en el desarrollo, convulsiones, cataratas, cardiomiopatía y anomalías neuromusculares; siendo estas últimas sólo descritas en unos pocos informes de casos recientes . El síndrome de Vici fue descrito por primera vez en 1988 por Dionisi Vici, et al. en el caso de dos hermanos que presentaban albinismo, ACC, cataratas, cardiomiopatía, retraso mental e inmunodeficiencia, y posteriormente fue denominado «síndrome de Vici» por del Campo, et al. en 1999. El síndrome de Vici se ha observado tanto en hombres como en mujeres, y se han identificado varias mutaciones relacionadas en el gen EPG5.
Los padres de nuestros pacientes tienen consanguinidad y apoyan la herencia autosómica recesiva del síndrome de Vici. Trece de los 31 pacientes, incluido el nuestro, son hermanos. Se han descrito casos en los que los padres eran primos lejanos.
El síndrome de Vici se debe a mutaciones recesivas en EPG5 en el cromosoma 18q12.3, que codifica la proteína 5 de los gránulos P ectópicos (EPG5), un regulador clave de la autofagia en los organismos superiores. La autofagia es una vía de degradación celular fundamental conservada a lo largo de la evolución, con importantes funciones en la eliminación de proteínas y orgánulos defectuosos, la defensa contra las infecciones y la adaptación a las cambiantes demandas metabólicas.
El desarrollo del SNC, la regulación inmunitaria y la pigmentación de la piel en pacientes con síndrome de Vici implican a la autofagia en una gama más amplia de procesos celulares. En el cerebro, la autofagia se ha investigado ampliamente como un mecanismo patogénico clave en varios trastornos neurodegenerativos .
La mayoría de los signos y síntomas del síndrome de Vici están presentes al nacer; algunos rasgos como las cataratas, la cardiomiopatía y la inmunodeficiencia no siempre están presentes al nacer, sino que se espera que evolucionen durante los primeros años . Y la mayoría de los pacientes han fallecido dentro de los tres primeros años de vida secundarios a cardiomiopatía o neumonía
Agradecimientos
Los autores desean dar las gracias a los padres del paciente y al laboratorio de diagnóstico saudí.
- Dionisi Vici C, Sabetta G, Gambarara M, Vigevano F, Bertini E, et al. (1988) Agenesia del cuerpo calloso, inmunodeficiencia combinada, catarata bilateral e hipopigmentación en dos hermanos. Am J Med Genet 29:1-8.
- Thomas Cullup, Ay Lin Kho, Carlo Dionisi-Vici (2013) Mutaciones recesivas en EPG5 causan el síndrome de Vici, un trastorno multisistémico con autofagia defectuosa. Nature Genetics 45: 83-87.
- Said E, Soler D, Sewry C (2012) Vici syndrome-a rapidly progressive neurodegenerative disorder with hypopigmentation, immunodeficiency and myopathic changes on muscle biopsy. Am J Med Genet A 158: 440-444.
- del Campo M, Hall BD, Aeby A, Nassogne MC, Verloes A, et al. (1999) Albinismo y agenesia del cuerpo calloso con profundo retraso del desarrollo: Síndrome de Vici, evidencia de herencia autosómica recesiva. Am J Med Genet 85: 479-485.
- Ehmke N, Parvaneh N, Krawitz P, Ashrafi MR, Karimi P, et al. (2014) Primera descripción de un paciente con síndrome de Vici debido a una mutación que afecta al penúltimo exón de EPG5 y revisión de la literatura. Am J Med Genet A 164A: 3170-3175.
- Al-Owain M, Al-Hashem A, Al-Muhaizea M, Humaidan H, Al-Hindi H, et al. (2010) Síndrome de Vici asociado a hipoplasia pulmonar unilateral y miopatía. Am J Med Genet A 152A: 1849-1853.
- Byrne S, Dionisi-Vici C, Smith L, Gautel M, Jungbluth H, et al. (2016) Síndrome de Vici: una revisión. Orphanet J Rare Dis 11: 21.
- Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, et al. (2006) Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol 76: 89-101.
- Cullup T, Dionisi-Vici C, Kho AL, Yau S, Mohammed S, et al. (2014) Tarjeta genética de utilidad clínica para: Síndrome de Vici. Eur J Hum Genet 22.
- Curtis Rogers R, Bridgette Aufmuth, Stephanie Monesson (2011) Vici Syndrome: Un raro síndrome autosómico recesivo con anomalías cerebrales, miocardiopatía y discapacidad intelectual severa. Informes de casos en genética 2011: 421582.