Take a look at the Recent articles
Abstract
Purpose: To report a case of oculocutaneous hypopigmentation found in an infant with sever hypotonia, cardiomyopathy and agenesis of the corpus.
Case Report: Deze studie betrof baby een man die werd geleverd na een zwangerschapsduur van 37 weken. Product van gezonde consanguine Yamani ouders met groeiachterstand, congenitale bilaterale cataract, en dysmorf gezicht, blond haar, en huid, nystagmus en hij had linker ventrikel hypertrofie. Mutaties in het gen epg5 zijn geïdentificeerd als de oorzaak van het Vici-syndroom.
Conclusie: Het Vici-syndroom is een duidelijke klinische entiteit. De belangrijkste klinische verschijnselen zijn hypotonai, ontwikkelingsachterstand, albinisme, cataract, cardiomyopathie, immuundeficiëntie, en agenese van het corpus callosum.
Key words
Vici syndroom; agenese van het corpus callosum; EPG5
Inleiding
Vici syndroom is een ernstige, recessief overervende congenitale aandoening die wordt gekenmerkt door de hoofdkenmerken van agenese van het corpus callosum (ACC), cataract, oculocutane hypopigmentatie, cardiomyopathie, failure to thrive en variabel.
Het werd voor het eerst beschreven in 1988, bij twee broers met een malformatiesyndroom bestaande uit agenese van het corpus callosum, cutane hypopigmentatie, bilaterale cataract, gespleten lip en gehemelte, en gecombineerde immunodeficiëntie. De broers en zussen leden aan ernstige psychomotorische retardatie, toevallen, recurrente ernstige infecties van de luchtwegen en chronische mucocutane candidiasis. Ze stierven aan bronchopneumonie op de leeftijd van 2 en 3 jaar.
Vici syndroom is een multisysteem aandoening geassocieerd met defecte autofagie en suggereert een fundamentele rol van de autofagie pathway in het immuunsysteem en de anatomische en functionele vorming van organen zoals de hersenen en het hart.
Clinical Report
Deze baby is het eerste product van gezonde consanguine Yamani Ouders. Hij was voldragen, na een ongestoorde zwangerschap met normale vertexbevalling bij 36 weken zwangerschapsduur met een geboortegewicht van 3,2 kg, een schedelomtrek van 31 cm en een Apgar-score van 8/8.
Toen hij 25 dagen oud was, werd hij in het ziekenhuis opgenomen met een voorgeschiedenis van hoest, kortademigheid en koorts gedurende de laatste drie dagen. Geassocieerd met cyanose, had de patiënt een geschiedenis van slecht zuigen en terugkerende verstikkingsaanvallen bij elke voeding sinds de geboorte, maar helaas werd dit verwaarloosd. Geen geschiedenis van braken of huiduitslag en geen geschiedenis van abnormale bewegingen. Geen familiegeschiedenis van abortussen of sterfgevallen.
Bij het onderzoek zag hij er bleek, cachectisch en dysmorf uit (figuur 1). (hypopegmented huid, oog, en blond haar, breed voorhoofd, hyperbolisme, lange philtrum en micrognathia). Zijn gewicht was 4.100 g (3-10e centiel), lengte 53 cm (3-10e centiel), en hoofdomtrek 33 cm (3e centiel). Ophthalmologie onderzoeken toonde milde horizontale nystagmus, bilaterale cataract en flair retina. Op de bilaterale borstkas was er veel crepitatie, geen hartruis met normaal hartgeluid en had diepe hypotonie.
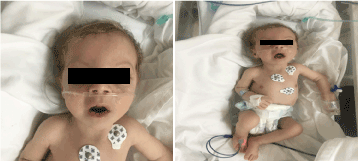
Figuur 1. Klinische foto van patiënt op de leeftijd van 2 maanden (a) Hypotonie, (b) blond haar lang philtrum en micrognathia
Laboratoriumonderzoek toonde microcytaire en hypochromische anemie, nierprofielen en coagulatie waren onopvallend. Alanine transaminase (ALT) en aspartaat aminotransferase (AST) waren verhoogd met respectievelijk 183-231 (10-45 U/L), en 193-301 (10-45 U/L). De bilirubine was normaal. CK: 389 U/L en LDH: 612 U/L. De ammoniak was normaal 13 mmol/L. De immunologische workup onthulde verlaagde immunoglobulinen niveaus IgA: 0,1 g/L (0,7-4), maar de IgG: 4,51 g/L (7-16), IgM: .8 g/L (0,4-2,3). Waren binnen de normale waarde. Lymfocyten subsets (CD3, CD4, CD8, CD19, en CD22) tellingen waren normaal. Bloed, urine en waren geen groei. De serologische test voor toxoplasma, rubella, cytomegalovirus, herpes simplex, en lever markers waren allemaal normaal. Röntgenfoto’s van de borst toonden pneumonische infiltratie en cardiomegalie. Abdominale ultrasonografie werd bilateraal hydronefrose onthuld. Linker ventrikel hypertrofie werd gedetecteerd door echocardiografisch onderzoek. Magnetic resonance imaging (MRI) van de hersenen bevestigde agenese van corpus callosum (figuur 2). Genetische testen werden bevestigd Een homozygote verlies van functie variant van EPG5 (EPG5:NM_020964:exon9:c.1924C>T:p.R642X)
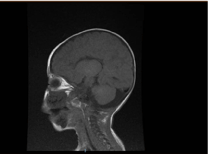
Figuur 2. MRI van de hersenen, Sagittale T1 gewogen sectie toont agenese van corpus callosum
Tijdens de 2e week van opname verslechterde zijn toestand snel met herhaalde episoden van aspiratiepneumonie die beademing vereisten. Uiteindelijk kreeg de patiënt een hartstilstand en overleed.
Materialen en methoden
WES wordt uitgevoerd op genomisch DNA met behulp van de Agilent Sure Select Target Enrichment-workflow voor het vastleggen van interessante regio’s uit een DNA-fragmentbibliotheek. Het volledige exoom wordt gesequeneerd op het Illumina HiSeq 2500 sequencing systeem met een minimale dekking van 30X van 95% van de doelregio’s. De DNA-sequenties van het exoom van de proband worden in kaart gebracht en vergeleken met de menselijke genoomsequentie UCSC hg19. Saudi Diagnostic Laboratories gebruikt een in-house pijplijn om de sequentie van de proband met de referentiesequentie te vergelijken. De dekking en kwaliteit van de coderende exonen van de bekende eiwitcoderende RefSeq-genen worden beoordeeld. Exoomanalyses ondervragen duizenden genetische varianten in een proband met behulp van eigen databases die zijn aangepast aan Arabische populaties. Subsets hiervan worden gekarakteriseerd aan de hand van de richtlijnen van het American College of Medical Genetics and Genomics (ACMG) (Richards, et al. 2015) om hun klinische relevantie te classificeren. Sanger sequencing is uitgevoerd om alle varianten die in het rapport zijn opgenomen te valideren, tenzij anders vermeld
Resultaten
Een homozygote loss of function-variant van EPG5 (EPG5:NM_020964:exon9:c.1924C>T:p.R642X) werd bij deze patiënt geïdentificeerd. Volgens de ACMG-richtlijnen (Gen in Med 17:5, 2015) moet deze worden geclassificeerd als een pathogene variant. Homozygote Loss of Function (LOF) varianten van EPG5 zijn een veel voorkomend mechanisme van het Vici-syndroom, 242840 .
Discussie
Vici syndroom, is een zeldzame congenitale multi-systeem aandoening die wordt overgeërfd in een autosomaal recessief patroon, het wordt gekenmerkt door falen om te gedijen, hypopigmentatie, immunologische defecten duidelijk door terugkerende infecties, neurologische afwijkingen van ontwikkelingsachterstand, toevallen, cataract, cardiomyopathie en neuromusculaire afwijkingen; met de latere alleen beschreven in een paar recente case reports . Het Vici-syndroom werd voor het eerst gemeld in 1988 door Dionisi Vici, et al. in het geval van twee broers met albinisme, ACC, cataract, cardiomyopathie, mentale retardatie, en immunodeficiëntie, en werd vervolgens ‘Vici-syndroom’ genoemd door del Campo, et al. in 1999. Vici syndroom is waargenomen bij zowel mannen als vrouwen, en een aantal gerelateerde mutaties zijn geïdentificeerd in het EPG5 gen .
Onze patiënt ouders hebben consanguiniteit en ondersteunen autosomaal recessieve overerving van het Vici syndroom. Dertien van de 31 patiënten, waaronder de onze, zijn broers en zussen. Er zijn gevallen gemeld waarbij de ouders verre neven en nichten waren.
Vici syndroom is het gevolg van recessieve mutaties in EPG5 op chromosoom 18q12.3, coderend voor ectopisch P granules proteïne 5 (EPG5), een belangrijke autofagie-regulator in hogere organismen. Autofagie is een fundamentele cellulaire degradatieroute die door de evolutie heen bewaard is gebleven en een belangrijke rol speelt bij de verwijdering van defecte eiwitten en organellen, de verdediging tegen infecties en de aanpassing aan veranderende metabolische eisen.
De ontwikkeling van het CZS, de immuunregulatie en de huidpigmentatie bij patiënten met het Vici-syndroom impliceren autofagie in een breder scala van cellulaire processen. In de hersenen is autofagie uitgebreid onderzocht als een belangrijk pathogeen mechanisme bij verschillende neurodegeneratieve aandoeningen.
De meeste tekenen en symptomen van het Vici-syndroom zijn bij de geboorte aanwezig; sommige kenmerken zoals cataract, cardiomyopathie en immunodeficiëntie zijn niet altijd bij de geboorte aanwezig, maar zullen zich naar verwachting in de loop van de eerste jaren ontwikkelen. En de meeste van de patiënten zijn overleden binnen de eerste drie jaar van het leven secundair aan cardiomyopathie of longontsteking
Acknowledgements
De auteurs willen de ouders van de patiënt en de Saoedische diagnostisch laboratorium bedanken.
- Dionisi Vici C, Sabetta G, Gambarara M, Vigevano F, Bertini E, et al. (1988) Agenese van het corpus callosum, gecombineerde immunodeficiëntie, bilaterale cataract, en hypopigmentatie bij twee broers. Am J Med Genet 29:1-8.
- Thomas Cullup, Ay Lin Kho, Carlo Dionisi-Vici (2013) Recessieve mutaties in EPG5 veroorzaken het Vici-syndroom, een multisysteemaandoening met defecte autofagie. Nature Genetics 45: 83-87.
- Said E, Soler D, Sewry C (2012) Vici-syndroom-een snel progressieve neurodegeneratieve aandoening met hypopigmentatie, immunodeficiëntie en myopathische veranderingen op spierbiopsie. Am J Med Genet A 158: 440-444.
- del Campo M, Hall BD, Aeby A, Nassogne MC, Verloes A, et al. (1999) Albinisme en agenese van het corpus callosum met een diepe ontwikkelingsachterstand: Vici-syndroom, bewijs voor autosomaal recessieve overerving. Am J Med Genet 85: 479-485.
- Ehmke N, Parvaneh N, Krawitz P, Ashrafi MR, Karimi P, et al. (2014) First description of a patient with Vici syndrome due to a mutation affecting the penultimate exon of EPG5 and review of the literature. Am J Med Genet A 164A: 3170-3175.
- Al-Owain M, Al-Hashem A, Al-Muhaizea M, Humaidan H, Al-Hindi H, et al. (2010) Vici-syndroom geassocieerd met unilaterale longhypoplasie en myopathie. Am J Med Genet A 152A: 1849-1853.
- Byrne S, Dionisi-Vici C, Smith L, Gautel M, Jungbluth H, et al. (2016) Vici-syndroom: een review. Orphanet J Rare Dis 11: 21.
- Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, et al. (2006) Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol 76: 89-101.
- Cullup T, Dionisi-Vici C, Kho AL, Yau S, Mohammed S, et al. (2014) Clinical utility gene card for: Vici-syndroom. Eur J Hum Genet 22.
- Curtis Rogers R, Bridgette Aufmuth, Stephanie Monesson (2011) Vici Syndrome: A Rare Autosomal Recessive Syndrome with Brain Anomalies, Cardiomyopathy, and Severe Intellectual Disability. Case Reports in Genetics 2011: 421582.