Se de senaste artiklarna
Abstract
Syfte: Att rapportera ett fall av okulokutan hypopigmentering som hittades hos ett spädbarn med allvarlig hypotoni, kardiomyopati och agenesi av corpus.
Fallrapport: Den här studien gällde ett spädbarn en man som föddes efter en graviditetsperiod på 37 veckor. Produkt av friska samkönade Yamani-föräldrar med tillväxthämning, medfödd bilateral grå starr och dysmorfisk ansiktsform, ljust hår och hud, nystagmus och han hade vänster ventrikelhypertrofi. Mutationer i genen epg5 har identifierats som orsak till Vici-syndromet.
Slutsats: Vici syndrom är en distinkt klinisk enhet. Dess huvudsakliga kliniska manifestationer omfattade hypotonai, utvecklingsförsening, albinism, katarakt, kardiomyopati, immunbrist och agenesi av corpus callosum.
Nyckelord
Vici syndrom; agenesi av corpus callosum; EPG5
Introduktion
Vici syndrom är en allvarlig, recessivt nedärvd medfödd sjukdom som kännetecknas av huvuddragen agenesi av corpus callosum (ACC), grå starr, okulokutan hypopigmentering, kardiomyopati, bristande tillväxt och varierande.
Den beskrevs första gången 1988, hos två bröder med ett missbildningssyndrom bestående av agenesi av corpus callosum, kutan hypopigmentering, bilateral katarakt, läpp- och gomspalt och kombinerad immunbrist. Syskonen led av allvarlig psykomotorisk retardation, kramper, återkommande svåra luftvägsinfektioner och kronisk mukokutan candidiasis. De dog av bronkopneumoni vid 2 och 3 års ålder .
Vici syndrom är en multisystemsjukdom som är förknippad med defekt autofagi och tyder på en grundläggande roll för autofagivägen i immunsystemet och den anatomiska och funktionella bildningen av organ som hjärnan och hjärtat .
Klinisk rapport
Denna bebis är den första produkten av friska samkönade Yamani-föräldrar. Han var fullgången, efter en händelselös graviditet med normal vertexförlossning vid 36 veckors gestationsålder med födelsevikt 3,2 kg, en kranieomkrets på 31 cm och en Apgarpoäng på 8/8.
Vid 25 dagars ålder blev han inlagd på sjukhus med anamnesen om hosta, andnöd och feber under de senaste tre dagarna. I samband med cyanos hade patienten en historia av dålig sugförmåga och återkommande kvävningsattacker vid varje matning sedan födseln, men tyvärr försummades han. Ingen historia av kräkningar eller hudutslag och ingen historia av onormala rörelser. Ingen familjehistoria med aborter eller dödsfall.
Vid undersökningen såg han blek, kachektisk och dysmorfisk ut (figur 1). (Hypopegmenterad hud, öga och blont hår, bred panna, hyperbolism, långt philtrum och mikrognathi). Hans vikt var 4 100 g (3-10:e centilen), längd 53 cm (3-10:e centilen) och huvudomkrets 33 cm (3:e centilen). Vid oftalmologiska undersökningar visades mild horisontell nystagmus, bilateral katarakt och flairretina. Full av crepitation på bilaterl bröstkorg, inget blåsljud med normalt hjärtljud och hade djup hypotoni.
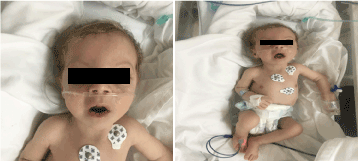
Figur 1. Klinisk bild av patienten vid 2 månaders ålder (a) Hypotoni, (b) blont hår långt philtrum och mikrognathi
Laboratorisk undersökning visade mikrocytisk och hypokrom anemi, njurprofiler och koagulation var okej. Alanintransaminas (ALT) och aspartataminotransferas (AST) var förhöjda med 183-231 (10-45 U/L) respektive 193-301 (10-45 U/L). Bilirubinet var normalt. CK: 389 U/L och LDH: 612 U/L. Ammoniaken var normal 13 mmol/L. Den immunologiska utredningen visade minskade immunoglobulinnivåer IgA: 0,1 g/L (0,7-4), men IgG: 4,51 g/L (7-16), IgM: 0,8 g/L (0,4-2,3). Var inom det normala värdet. Antalet undergrupper av lymfocyter (CD3, CD4, CD8, CD19 och CD22) var normalt. Blod, urin och var ingen tillväxt. Det serologiska testet för toxoplasma, röda hund, cytomegalovirus, herpes simplex och hepatiska markörer var alla normala. Röntgen av bröstkorgen visade pneumonisk infiltration och kardiomegali. Abdominal ultraljud visade bilateral hydronefrosis. Vänsterkammarhypertrofi påvisades genom ekokardiografisk undersökning. Magnetisk resonanstomografi (MRT) av hjärnan bekräftade agenesi av corpus callosum (figur 2). Genetisk testning bekräftade En homozygot funktionsförlustvariant av EPG5 (EPG5:NM_020964:exon9:c.1924C>T:p.R642X)
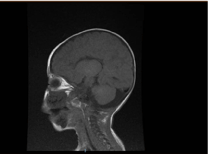
Figur 2. MRT av hjärnan, Sagittalt T1-vägt snitt som visar agenesi av corpus callosum
Under den andra veckan av intagningen försämrades hans tillstånd snabbt med upprepade episoder av aspirationspneumoni som krävde ventilation. Så småningom fick patienten hjärt- och lungstillestånd och avled.
Material och metoder
WES utförs på genomiskt DNA med hjälp av arbetsflödet Agilent Sure Select Target Enrichment för att fånga regioner av intresse från ett DNA-fragmentbibliotek. Hela exomet sekvenseras på Illumina HiSeq 2500-sekvenseringssystemet med en minsta täckning på 30X av 95 % av målregionerna. Probandens exom-DNA-sekvenser kartläggs och jämförs med UCSC hg19-referenssekvensen från det mänskliga genomet. Saudi Diagnostic Laboratories använder en egen pipeline för att jämföra probandens sekvens med referenssekvensen. Bedömning av täckning och kvalitet för riktade kodande exoner i de kända proteinkodande RefSeq-generna utförs. Exomanalyser undersöker tusentals genetiska varianter hos en försöksperson med hjälp av egenutvecklade databaser som är anpassade till arabiska populationer. Undergrupper av dessa karakteriseras med hjälp av riktlinjer från American College of Medical Genetics and Genomics (ACMG) (Richards, et al. 2015) för att klassificera deras kliniska relevans. Sangersekvensering utförs för att validera alla varianter som ingår i rapporten om inget annat anges
Resultat
En homozygot funktionsförlustvariant av EPG5 (EPG5:NM_020964:exon9:c.1924C>T:p.R642X) identifierades hos denna patient. Enligt ACMG:s riktlinjer (Gen in Med 17:5, 2015) ska den klassificeras som en patogen variant. Homozygota varianter av funktionsförlust (LOF) av EPG5 är en vanlig mekanism för Vici-syndromet, 242840 .
Diskussion
Vici syndrom, är en sällsynt medfödd multisystemstörning som ärvs i ett autosomalt recessivt mönster, och kännetecknas av bristande tillväxt, hypopigmentering, immunologiska defekter som visar sig i form av återkommande infektioner, neurologiska avvikelser i form av utvecklingsfördröjning, kramper, grå starr, kardiomyopati och neuromuskulära avvikelser; med det senare endast beskrivet i några få nyligen publicerade fallrapporter . Vici-syndromet rapporterades för första gången 1988 av Dionisi Vici, et al. om två bröder med albinism, ACC, grå starr, kardiomyopati, mental retardation och immunbrist, och fick därefter namnet ”Vici-syndromet” av del Campo, et al. 1999. Vici syndrom har observerats hos både män och kvinnor, och ett antal relaterade mutationer har identifierats i EPG5-genen .
Våra patientföräldrar är släkt med varandra och stöder autosomal recessiv nedärvning av Vici syndrom. Tretton av de 31 patienterna, inklusive vår, är syskon. Fall har rapporterats där föräldrarna var avlägsna kusiner .
Vici syndrom beror på recessiva mutationer i EPG5 på kromosom 18q12.3, som kodar för ektopiskt P-granulatprotein 5 (EPG5), en viktig autofagireglerare i högre organismer. Autofagi är en grundläggande cellulär nedbrytningsväg som bevarats genom hela evolutionen med viktiga roller i avlägsnandet av defekta proteiner och organeller, försvar mot infektioner och anpassning till förändrade metaboliska krav.
CNSutveckling, immunreglering och hudpigmentering hos patienter med Vici-syndromet innebär att autofagi är inblandat i ett bredare spektrum av cellulära processer. I hjärnan har autofagi undersökts ingående som en viktig patogen mekanism vid olika neurodegenerativa sjukdomar .
De flesta tecken och symtom på Vici-syndromet finns vid födseln; vissa egenskaper som katarakt, kardiomyopati och immunbrist finns inte alltid vid födseln, utan förväntas utvecklas under de första åren . Och de flesta av patienterna har dött inom de tre första levnadsåren sekundärt till kardiomyopati eller lunginflammation
Acknowledgements
Författarna vill tacka föräldrarna till patienten och Saudiarabiens diagnostiska laboratorium.
- Dionisi Vici C, Sabetta G, Gambarara M, Vigevano F, Bertini E, et al. (1988) Agenesi av corpus callosum, kombinerad immunbrist, bilateral katarakt och hypopigmentering hos två bröder. Am J Med Genet 29:1-8.
- Thomas Cullup, Ay Lin Kho, Carlo Dionisi-Vici (2013) Recessiva mutationer i EPG5 orsakar Vici syndrom, en multisystemsjukdom med defekt autofagi. Nature Genetics 45: 83-87.
- Said E, Soler D, Sewry C (2012) Vici syndrom – en snabbt progressiv neurodegenerativ sjukdom med hypopigmentering, immunbrist och myopatiska förändringar vid muskelbiopsi. Am J Med Genet A 158: 440-444.
- del Campo M, Hall BD, Aeby A, Nassogne MC, Verloes A, et al. (1999) Albinism och agenesi av corpus callosum med djup utvecklingsförsening: Vici syndrom, bevis för autosomalt recessivt arv. Am J Med Genet 85: 479-485.
- Ehmke N, Parvaneh N, Krawitz P, Ashrafi MR, Karimi P, et al. (2014) Första beskrivningen av en patient med Vici syndrom på grund av en mutation som påverkar det näst sista exonet av EPG5 och genomgång av litteraturen. Am J Med Genet A 164A: 3170-3175.
- Al-Owain M, Al-Hashem A, Al-Muhaizea M, Humaidan H, Al-Hindi H, et al. (2010) Vici syndrom associerat med unilateral lunghypoplasi och myopati. Am J Med Genet A 152A: 1849-1853.
- Byrne S, Dionisi-Vici C, Smith L, Gautel M, Jungbluth H, et al. (2016) Vici syndrome: a review. Orphanet J Rare Dis 11: 21.
- Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, et al. (2006) Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol 76: 89-101.
- Cullup T, Dionisi-Vici C, Kho AL, Yau S, Mohammed S, et al. (2014) Clinical utility gene card for: Vici syndrom. Eur J Hum Genet 22.
- Curtis Rogers R, Bridgette Aufmuth, Stephanie Monesson (2011) Vici syndrom: Ett sällsynt autosomalt recessivt syndrom med hjärnanomalier, kardiomyopati och allvarlig intellektuell funktionsnedsättning. Case Reports in Genetics 2011: 421582.