Take a look at the Recent articles
Abstract
Purpose: To report a case of oculocutaneous hypopigmentation found in an infant with sever hypotonia, cardiomyopathy and agenesis of the corpus.
Case Report: Ez a vizsgálat egy férfi csecsemőt érintett, aki 37 hetes terhességi idő után jött világra. Egészséges vérszerinti yamani szülők gyermeke, növekedési elmaradással, veleszületett kétoldali szürkehályoggal és diszmorf arccal, világos hajjal és bőrrel, nystagmussal, valamint bal kamrai hipertrófiával. A Vici-szindróma okaként az epg5 gén mutációit azonosították.
Következtetés: A Vici-szindróma egy különálló klinikai entitás. Fő klinikai megnyilvánulásai közé tartozik a hypotonai, a fejlődési késés, az albinizmus, a szürkehályog, a cardiomyopathia, az immunhiány és a corpus callosum agenezise.
Kulcsszavak
Vici-szindróma; corpus callosum agenezise; EPG5
Bevezetés
A Vici-szindróma egy súlyos, recesszíven öröklődő veleszületett rendellenesség, melynek fő jellemzői a corpus callosum (ACC) agenezise, a szürkehályog, az oculocutan hipopigmentáció, a cardiomyopathia, a gyarapodás elmaradása és a változó.
Először 1988-ban írták le két testvérnél, akiknél a corpus callosum ageneziséből, bőr hipopigmentációból, kétoldali szürkehályogból, ajak- és szájpadhasadékból, valamint kombinált immunhiányból álló malformációs szindrómát észleltek. A testvérek súlyos pszichomotoros retardációban, görcsrohamokban, visszatérő súlyos légúti fertőzésekben és krónikus nyálkahártya candidiasisban szenvedtek. Hörgőtüdőgyulladásban haltak meg 2 és 3 éves korukban.
A Vici-szindróma egy több rendszerre kiterjedő rendellenesség, amely hibás autofágiával jár, és az autofágia útvonal alapvető szerepére utal az immunrendszerben és az olyan szervek, mint az agy és a szív anatómiai és funkcionális kialakulásában.
Klinikai jelentés
Ez a baba az egészséges vérszerinti Yamani szülők első gyermeke. Eseménytelen terhesség után, 36 hetes terhességi korban, normál csigolyaszüléssel, 3,2 kg-os születési súllyal, 31 cm-es koponyakörfogattal és 8/8-as Apgar pontszámmal született.
25 napos korában kórházba került köhögéssel, légszomjjal és lázzal az elmúlt három napban. Cianózissal társult, a beteg anamnézisében születése óta rossz szopási és minden etetésnél visszatérő fulladási rohamok szerepeltek, de sajnos elhanyagolták. Hányás, bőrkiütés és kóros mozgás nem volt a kórtörténetben. Családjában nem volt abortusz vagy haláleset.
A vizsgálat során sápadtnak, kachektikusnak és diszmorfnak tűnt (1. ábra). (hypopegmentált bőr, szem és szőke haj, széles homlok, hyperbolismus,hosszú philtrum és micrognathia). Súlya 4100 g volt (3-10. centilis), hossza 53 cm (3-10. centilis), fejkörfogata 33 cm (3. centilis). A szemészeti vizsgálatok enyhe horizontális nystagmust, kétoldali szürkehályogot és flair retinát mutattak. A kétoldali mellkason teljes krepitáció, normális szívhanggal, zörej nélkül, és mély hipotónia volt.
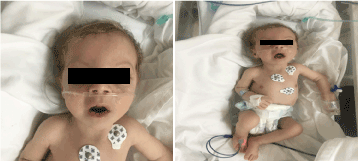
1. ábra. A beteg klinikai képe 2 hónapos korában (a) hipotónia, (b) szőke haj, hosszú philtrum és mikrognathia
A laboratóriumi vizsgálatok mikrocitikus és hipokróm vérszegénységet mutattak, a vese profilok és a véralvadás rendben voltak. Az alanin-transzamináz (ALT) és az aszpartát-aminotranszferáz (AST) értéke 183-231 (10-45 U/L), illetve 193-301 (10-45 U/L) volt emelkedett. A bilirubin normális volt. CK: 389 U/L és LDH: 612 U/L. Az ammónia normális volt 13 mmol/L. Az immunológiai vizsgálat csökkent immunglobulinszintet mutatott IgA: 0,1 g/L (0,7-4), de az IgG: 4,51 g/L (7-16), IgM: 0,8 g/L (0,4-2,3). A normál értéken belül voltak. A limfocita alcsoportok (CD3, CD4, CD8, CD19 és CD22) száma normális volt. Vér, vizelet és nem volt növekedés. A toxoplazma, rubeola, citomegalovírus, herpes simplex és a májmarkerek szerológiai vizsgálata normális volt. A mellkasröntgen tüdőinfiltrációt és cardiomegáliát mutatott. A hasi ultrahangvizsgálat kétoldali hydronephrosist mutatott. Echokardiográfiás vizsgálattal bal kamrai hipertrófiát észleltek. Az agy mágneses rezonanciás képalkotása (MRI) megerősítette a corpus callosum agenezisét (2. ábra). A genetikai vizsgálat megerősítette az EPG5 homozigóta funkcióvesztéses variánsát (EPG5:NM_020964:exon9:c.1924C>T:p.R642X)
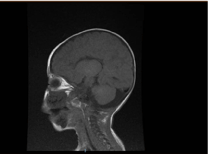
2. ábra. Az agy MRI-je, Sagittalis T1 súlyozott metszet, amely a corpus callosum agenezisét mutatja
A felvétel 2. hetében állapota gyorsan romlott, ismételt aspirációs tüdőgyulladásos epizódokkal, amelyek lélegeztetést igényeltek. Végül a beteg kardiopulmonális leállást szenvedett és meghalt.
Anyagok és módszerek
A WES-t genomiális DNS-en végezzük az Agilent Sure Select Target Enrichment munkafolyamat segítségével a DNS-fragmentkönyvtárból az érdeklődésre számot tartó régiók rögzítésére. A teljes exom szekvenálását az Illumina HiSeq 2500 szekvenáló rendszerrel végezzük, a célrégiók 95%-ának legalább 30X-os lefedettségével. A probandus exom DNS-szekvenciáit feltérképezik és összehasonlítják a humán genomot felépítő UCSC hg19 referenciaszekvenciával. A Saudi Diagnostic Laboratories egy házon belüli csővezetéket használ a vizsgálati személy szekvenciájának a referenciaszekvenciával való összehasonlítására. Az ismert fehérjekódoló RefSeq gének célzott kódoló exonjainak lefedettségét és minőségét értékelik. Az exom-elemzések több ezer genetikai variánst kérdeznek le a vizsgálati személyben az arab populációkra szabott, saját fejlesztésű adatbázisok segítségével. Ezek részhalmazait az American College of Medical Genetics and Genomics (ACMG) (Richards, et al. 2015) iránymutatásai alapján jellemzik, hogy osztályozzák klinikai relevanciájukat. A jelentésben szereplő összes variáns validálására Sanger-szekvenálást végeztünk, hacsak másként nem szerepel
Eredmények
Ebben a betegben az EPG5 homozigóta funkcióvesztéses variánsát (EPG5:NM_020964:exon9:c.1924C>T:p.R642X) azonosították. Az ACMG irányelvei szerint (Gen in Med 17:5, 2015) patogén variánsnak kell minősíteni. Az EPG5 homozigóta funkcióvesztéses (LOF) variánsai a Vici-szindróma gyakori mechanizmusa, 242840 .
Diszkusszió
A Vici szindróma, egy ritka veleszületett multiszisztémás rendellenesség, amely autoszomális recesszív módon öröklődik, jellemzője a gyarapodás elmaradása, hipopigmentáció, visszatérő fertőzésekkel nyilvánvaló immunológiai rendellenességek, neurológiai rendellenességek a fejlődési késés, görcsök, szürkehályog, kardiomiopátia és neuromuszkuláris rendellenességek; az utóbbiakat csak néhány újabb esetleírásban írták le . A Vici-szindrómáról először 1988-ban Dionisi Vici és munkatársai számoltak be két testvér esetében, akiknél albinizmus, ACC, szürkehályog, kardiomiopátia, mentális retardáció és immunhiány fordult elő, majd 1999-ben del Campo és munkatársai “Vici-szindrómának” nevezték el. A Vici-szindrómát férfiaknál és nőknél egyaránt megfigyelték, és számos kapcsolódó mutációt azonosítottak az EPG5 génben .
Páciensünk szülei vérségi rokonságban állnak egymással, ami alátámasztja a Vici-szindróma autoszomális recesszív öröklődését. A 31 beteg közül tizenhárom, köztük a miénk is, testvérpár. Olyan esetekről is beszámoltak, amelyekben a szülők távoli unokatestvérek voltak .
A Vici-szindróma a 18q12.3 kromoszómán található EPG5 recesszív mutációjának következménye, amely a magasabb szervezetekben az autofágia kulcsfontosságú szabályozóját, az ectopic P granules protein 5-öt (EPG5) kódolja. Az autofágia az evolúció során konzerválódott alapvető sejtes lebontó útvonal, amely fontos szerepet játszik a hibás fehérjék és organellumok eltávolításában, a fertőzések elleni védekezésben és a változó metabolikus igényekhez való alkalmazkodásban.
A Vici-szindrómás betegeknél az autofágia a sejtfolyamatok szélesebb körében játszik szerepet. Az agyban az autofágiát széles körben vizsgálták, mint a különböző neurodegeneratív rendellenességek kulcsfontosságú patogén mechanizmusát .
A vici szindróma legtöbb jele és tünete születéskor jelen van; néhány jellemző, mint a szürkehályog, a kardiomiopátia és az immunhiány nem mindig van jelen születéskor, hanem várhatóan az első évek során alakul ki . A legtöbb beteg pedig az élet első három évében halt meg másodlagosan kardiomiopátia vagy tüdőgyulladás következtében
Köszönet
A szerzők köszönetet mondanak a beteg szüleinek és a szaúdi diagnosztikai laboratóriumnak.
- Dionisi Vici C, Sabetta G, Gambarara M, Vigevano F, Bertini E, et al. (1988) Agenesis of the corpus callosum, combined immunodeficiency, bilateral cataract, and hypopigmentation in two brothers. Am J Med Genet 29:1-8.
- Thomas Cullup, Ay Lin Kho, Carlo Dionisi-Vici (2013) Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nature Genetics 45: 83-87.
- Said E, Soler D, Sewry C (2012) Vici syndrome-a rapidly progressive neurodegenerative disorder with hypopigmentation, immunodeficiency and myopathic changes on muscle biopsy. Am J Med Genet A 158: 440-444.
- del Campo M, Hall BD, Aeby A, Nassogne MC, Verloes A, et al. (1999) Albinism and agenesis of the corpus callosum with profound developmental delay: Vici-szindróma, az autoszomális recesszív öröklődés bizonyítéka. Am J Med Genet 85: 479-485.
- Ehmke N, Parvaneh N, Krawitz P, Ashrafi MR, Karimi P, et al. (2014) First description of a patient with Vici syndrome due to a mutation affecting the penultimate exon of EPG5 and review of the literature. Am J Med Genet A 164A: 3170-3175.
- Al-Owain M, Al-Hashem A, Al-Muhaizea M, Humaidan H, Al-Hindi H, et al. (2010) Egyoldali tüdőhipoplasiával és myopathiával társuló Vici-szindróma. Am J Med Genet A 152A: 1849-1853.
- Byrne S, Dionisi-Vici C, Smith L, Gautel M, Jungbluth H, et al. (2016) Vici syndrome: a review. Orphanet J Rare Dis 11: 21.
- Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, et al. (2006) Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol 76: 89-101.
- Cullup T, Dionisi-Vici C, Kho AL, Yau S, Mohammed S, et al. (2014) Clinical utility gene card for: Vici szindróma. Eur J Hum Genet 22.
- Curtis Rogers R, Bridgette Aufmuth, Stephanie Monesson (2011) Vici Syndrome: A Rare Autosomal Recessive Syndrome with Brain Anomalies, Cardiomyopathy, and Severe Intellectual Disability. Case Reports in Genetics 2011: 421582.