Sehen Sie sich die neuesten Artikel an
Abstract
Zweck: Bericht über einen Fall von okulokutaner Hypopigmentierung bei einem Säugling mit schwerer Hypotonie, Kardiomyopathie und Agenesie des Corpus
Fallbericht: Bei dieser Studie handelte es sich um ein männliches Baby, das nach einer Gestationszeit von 37 Wochen entbunden wurde. Das Kind gesunder konsanguiner Yamani-Eltern wies eine Wachstumsverzögerung, kongenitale beidseitige Katarakte, dysmorphe Gesichtszüge, helle Haare und Haut, Nystagmus und eine Hypertrophie des linken Ventrikels auf. Mutationen im Gen epg5 wurden als Ursache des Vici-Syndroms identifiziert.
Schlussfolgerung: Das Vici-Syndrom ist eine eigenständige klinische Entität. Zu seinen wichtigsten klinischen Manifestationen gehören Hypotonai, Entwicklungsverzögerung, Albinismus, Katarakt, Kardiomyopathie, Immunschwäche und Agenesie des Corpus callosum.
Schlüsselwörter
Vici-Syndrom; Agenesie des Corpus callosum; EPG5
Einführung
Das Vici-Syndrom ist eine schwere, rezessiv vererbte angeborene Störung, die durch die Hauptmerkmale Agenesie des Corpus callosum (ACC), Katarakte, okulokutane Hypopigmentierung, Kardiomyopathie, Gedeihstörung und Variabilität gekennzeichnet ist.
Erstmals beschrieben wurde sie 1988 bei zwei Brüdern mit einem Fehlbildungssyndrom, das aus einer Agenesie des Corpus callosum, einer kutanen Hypopigmentierung, einer bilateralen Katarakt, einer Lippen-Kiefer-Gaumenspalte und einer kombinierten Immunschwäche bestand. Die Geschwister litten unter schwerer psychomotorischer Retardierung, Krampfanfällen, wiederkehrenden schweren Atemwegsinfektionen und chronischer mukokutaner Candidose. Sie starben im Alter von 2 und 3 Jahren an Bronchopneumonie.
Das Vici-Syndrom ist eine Multisystemstörung, die mit einer gestörten Autophagie einhergeht und auf eine grundlegende Rolle des Autophagie-Stoffwechsels im Immunsystem und bei der anatomischen und funktionellen Bildung von Organen wie Gehirn und Herz hindeutet.
Klinischer Bericht
Dieses Baby ist das erste Produkt gesunder konsanguiner Yamani-Eltern. Nach einer ereignislosen Schwangerschaft mit normaler Scheitelentbindung in der 36. Schwangerschaftswoche, einem Geburtsgewicht von 3,2 kg, einem Schädelumfang von 31 cm und einem Apgar-Score von 8/8 war er voll ausgetragen.
Im Alter von 25 Tagen wurde er mit Husten, Kurzatmigkeit und Fieber für die letzten drei Tage ins Krankenhaus eingeliefert. In Verbindung mit Zyanose hatte der Patient seit seiner Geburt eine schlechte Saugleistung und wiederkehrende Erstickungsanfälle bei jeder Fütterung, wurde aber leider vernachlässigt. Keine Anamnese von Erbrechen oder Hautausschlag und keine Anamnese von abnormalen Bewegungen. In der Familienanamnese gab es keine Fehlgeburten oder Todesfälle.
Bei der Untersuchung wirkte er blass, kachektisch und dysmorph (Abbildung 1). (hypopegmentierte Haut, Augen und blondes Haar, breite Stirn, Hyperbolismus, langes Philtrum und Mikrognathie). Sein Gewicht betrug 4.100 g (3-10. Perzentile), seine Körperlänge 53 cm (3-10. Perzentile) und sein Kopfumfang 33 cm (3. Perzentile). Die augenärztliche Untersuchung ergab einen leichten horizontalen Nystagmus, einen beidseitigen Katarakt und eine flair retina. Der Brustkorb war voll mit Krepitation, kein Herzgeräusch mit normalen Herztönen und hatte eine ausgeprägte Hypotonie.
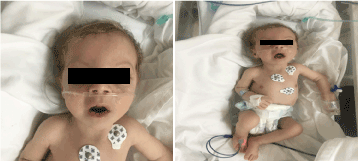
Abbildung 1. Klinisches Foto der Patientin im Alter von 2 Monaten (a) Hypotonie, (b) blondes Haar, langes Philtrum und Mikrognathie
Die Laboruntersuchung ergab eine mikrozytäre und hypochrome Anämie, Nierenprofile und Gerinnung waren unauffällig. Die Alanin-Transaminase (ALT) und die Aspartat-Aminotransferase (AST) waren mit 183-231 (10-45 U/L) bzw. 193-301 (10-45 U/L) erhöht. Das Bilirubin war normal. CK: 389 U/L und LDH: 612 U/L. Der Ammoniakwert war mit 13 mmol/L normal. Die immunologische Untersuchung ergab reduzierte Immunglobulinwerte IgA: 0,1 g/L (0,7-4), aber IgG: 4,51 g/L (7-16), IgM: .8 g/L (0,4-2,3). lagen innerhalb des Normalwerts. Die Anzahl der Lymphozyten-Untergruppen (CD3, CD4, CD8, CD19 und CD22) war normal. Blut, Urin und waren kein Wachstum. Die serologischen Tests auf Toxoplasma, Röteln, Cytomegalovirus, Herpes simplex und Lebermarker waren alle normal. Die Röntgenaufnahme des Brustkorbs zeigte eine Lungeninfiltration und eine Kardiomegalie. Bei der Abdomensonographie wurde eine beidseitige Hydronephrose festgestellt. In einer echokardiographischen Untersuchung wurde eine linksventrikuläre Hypertrophie festgestellt. Die Magnetresonanztomographie (MRT) des Gehirns bestätigte eine Agenesie des Corpus callosum (Abbildung 2). Ein Gentest bestätigte eine homozygote Funktionsverlustvariante von EPG5 (EPG5:NM_020964:exon9:c.1924C>T:p.R642X)
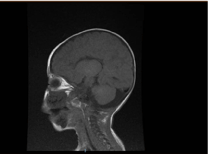
Abbildung 2. MRT des Gehirns, sagittaler T1-gewichteter Schnitt, der eine Agenesie des Corpus callosum zeigt
In der zweiten Woche nach der Einlieferung verschlechterte sich sein Zustand schnell mit wiederholten Episoden einer Aspirationspneumonie, die eine Beatmung erforderlich machte. Schließlich erlitt der Patient einen Herz-Lungen-Stillstand und starb.
Materialien und Methoden
Die WES wird an genomischer DNA unter Verwendung des Agilent Sure Select Target Enrichment Workflows durchgeführt, um interessante Regionen aus einer DNA-Fragmentbibliothek zu erfassen. Das gesamte Exom wird auf dem Illumina HiSeq 2500 Sequenziersystem sequenziert, mit einer Mindestabdeckung von 30X von 95% der Zielregionen. Die Exom-DNA-Sequenzen des Probanden werden kartiert und mit der Referenzsequenz des menschlichen Genoms (UCSC hg19) verglichen. Saudi Diagnostic Laboratories vergleicht die Sequenz des Probanden mit der Referenzsequenz mithilfe einer internen Pipeline. Es wird eine Bewertung der Abdeckung und der Qualität für gezielte kodierende Exons der bekannten protein-kodierenden RefSeq-Gene durchgeführt. Bei Exomanalysen werden Tausende von genetischen Varianten eines Probanden mit Hilfe proprietärer, auf arabische Populationen zugeschnittener Datenbanken abgefragt. Teilmengen davon werden anhand der Richtlinien des American College of Medical Genetics and Genomics (ACMG) (Richards, et al. 2015) charakterisiert, um ihre klinische Relevanz zu klassifizieren. Sofern nicht anders angegeben, wurden alle im Bericht enthaltenen Varianten durch Sanger-Sequenzierung validiert
Ergebnisse
Bei diesem Patienten wurde eine homozygote Funktionsverlustvariante von EPG5 (EPG5:NM_020964:exon9:c.1924C>T:p.R642X) identifiziert. Gemäß den ACMG-Richtlinien (Gen in Med 17:5, 2015) sollte sie als pathogene Variante eingestuft werden. Homozygote Loss of Function (LOF)-Varianten von EPG5 sind ein häufiger Mechanismus des Vici-Syndroms, 242840 .
Diskussion
Das Vici-Syndrom ist eine seltene angeborene Multisystemstörung, die autosomal rezessiv vererbt wird und durch Gedeihstörung, Hypopigmentierung, immunologische Defekte, die sich durch wiederkehrende Infektionen zeigen, neurologische Anomalien wie Entwicklungsverzögerung, Krampfanfälle, Katarakte, Kardiomyopathie und neuromuskuläre Anomalien gekennzeichnet ist, wobei letztere nur in einigen wenigen aktuellen Fallberichten beschrieben wurden. Das Vici-Syndrom wurde erstmals 1988 von Dionisi Vici et al. bei zwei Brüdern mit Albinismus, ACC, Katarakt, Kardiomyopathie, geistiger Retardierung und Immunschwäche beschrieben und anschließend von del Campo et al. 1999 als „Vici-Syndrom“ bezeichnet. Das Vici-Syndrom wurde sowohl bei Männern als auch bei Frauen beobachtet, und im EPG5-Gen wurde eine Reihe verwandter Mutationen identifiziert.
Unsere Patienteneltern sind blutsverwandt und unterstützen die autosomal-rezessive Vererbung des Vici-Syndroms. Dreizehn der 31 Patienten, einschließlich unseres Patienten, sind Geschwister. Es wurden Fälle berichtet, in denen die Eltern entfernte Cousins waren.
Das Vici-Syndrom wird durch rezessive Mutationen in EPG5 auf Chromosom 18q12.3 verursacht, das für das ektopische P-Granulatprotein 5 (EPG5) kodiert, einen wichtigen Autophagie-Regulator in höheren Organismen. Die Autophagie ist ein grundlegender zellulärer Abbauweg, der im Laufe der Evolution konserviert wurde und eine wichtige Rolle bei der Beseitigung defekter Proteine und Organellen, der Abwehr von Infektionen und der Anpassung an sich ändernde Stoffwechselbedürfnisse spielt.
Die Entwicklung des ZNS, die Immunregulation und die Hautpigmentierung bei Patienten mit Vici-Syndrom deuten darauf hin, dass die Autophagie an einer Vielzahl von Zellprozessen beteiligt ist. Im Gehirn wurde die Autophagie als ein wichtiger pathogener Mechanismus bei verschiedenen neurodegenerativen Erkrankungen eingehend untersucht.
Die meisten Anzeichen und Symptome des Vici-Syndroms sind bei der Geburt vorhanden; einige Merkmale wie Katarakte, Kardiomyopathie und Immundefizienz sind nicht immer bei der Geburt vorhanden, sondern entwickeln sich voraussichtlich in den ersten Jahren. Die meisten Patienten starben innerhalb der ersten drei Lebensjahre an Kardiomyopathie oder Lungenentzündung.
Danksagung
Die Autoren danken den Eltern der Patienten und dem saudischen Diagnoselabor.
- Dionisi Vici C, Sabetta G, Gambarara M, Vigevano F, Bertini E, et al. (1988) Agenesie des Corpus callosum, kombinierte Immunschwäche, bilateraler Katarakt und Hypopigmentierung bei zwei Brüdern. Am J Med Genet 29:1-8.
- Thomas Cullup, Ay Lin Kho, Carlo Dionisi-Vici (2013) Rezessive Mutationen in EPG5 verursachen das Vici-Syndrom, eine Multisystemstörung mit gestörter Autophagie. Nature Genetics 45: 83-87.
- Said E, Soler D, Sewry C (2012) Vici syndrome-a rapidly progressive neurodegenerative disorder with hypopigmentation, immunodeficiency and myopathic changes on muscle biopsy. Am J Med Genet A 158: 440-444.
- del Campo M, Hall BD, Aeby A, Nassogne MC, Verloes A, et al. (1999) Albinism and agenesis of the corpus callosum with profound developmental delay: Vici-Syndrom, Beweis für autosomal rezessiven Erbgang. Am J Med Genet 85: 479-485.
- Ehmke N, Parvaneh N, Krawitz P, Ashrafi MR, Karimi P, et al. (2014) First description of a patient with Vici syndrome due to a mutation affecting the penultimate exon of EPG5 and review of the literature. Am J Med Genet A 164A: 3170-3175.
- Al-Owain M, Al-Hashem A, Al-Muhaizea M, Humaidan H, Al-Hindi H, et al. (2010) Vici-Syndrom in Verbindung mit einseitiger Lungenhypoplasie und Myopathie. Am J Med Genet A 152A: 1849-1853.
- Byrne S, Dionisi-Vici C, Smith L, Gautel M, Jungbluth H, et al. (2016) Vici syndrome: a review. Orphanet J Rare Dis 11: 21.
- Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, et al. (2006) Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol 76: 89-101.
- Cullup T, Dionisi-Vici C, Kho AL, Yau S, Mohammed S, et al. (2014) Clinical utility gene card for: Vici Syndrome. Eur J Hum Genet 22.
- Curtis Rogers R, Bridgette Aufmuth, Stephanie Monesson (2011) Vici Syndrome: Ein seltenes autosomal-rezessives Syndrom mit Hirnanomalien, Kardiomyopathie und schwerer geistiger Behinderung. Case Reports in Genetics 2011: 421582.