Take a look at the Recent articles
Abstract
Scop: Pentru a raporta un caz de hipopigmentare oculocutanată găsită la un sugar cu hipotonie severă, cardiomiopatie și agenezie de corp.
Case Report: Acest studiu a implicat bebelușul un bărbat care a fost adus pe lume după o perioadă gestațională de 37 de săptămâni. Produs al unor părinți Yamani consangvini sănătoși, cu retard de creștere, cataractă bilaterală congenitală și dismorfie facială, păr și piele albă, nistagmus și a avut hipertrofie de ventricul stâng. Mutații în gena epg5 au fost identificate ca fiind cauza sindromului Vici.
Concluzie: Sindromul Vici este o entitate clinică distinctă. Principalele sale manifestări clinice au inclus hipotonie, întârziere în dezvoltare, albinism, cataractă, cardiomiopatie, imunodeficiență și agenezie a corpului calos.
Cuvintele cheie
Sindromul Vici; agenezie de corp calos; EPG5
Introducere
Sindromul Vici este o tulburare congenitală severă, moștenită în mod recesiv, caracterizată prin principalele trăsături de agenezie a corpului calos (ACC), cataractă, hipopigmentare oculocutanată, cardiomiopatie, insuficiență de dezvoltare și variabilă.
A fost descrisă pentru prima dată în 1988, la doi frați cu un sindrom malformativ constând în agenezie de corp calos, hipopigmentare cutanată, cataractă bilaterală, fisură labială și palatină și imunodeficiență combinată. Frații sufereau de retard psihomotor sever, convulsii, infecții respiratorii severe recurente și candidoză mucocutanată cronică. Au murit de bronhopneumonie la vârstele de 2 și 3 ani .
Sindromul Vici este o tulburare multisistemică asociată cu autofagia defectuoasă și sugerează un rol fundamental al căii autofagiei în sistemul imunitar și în formarea anatomică și funcțională a unor organe precum creierul și inima .
Raport clinic
Acest copil este primul produs al părinților Yamani consangvini sănătoși. A fost născut la termen, după o sarcină fără evenimente, cu o naștere normală pe vertex la vârsta gestațională de 36 de săptămâni, cu o greutate la naștere de 3,2 kg, o circumferință craniană de 31 cm și un scor Apgar de 8/8.
La vârsta de 25 de zile, a fost spitalizat cu antecedente de tuse, dificultăți de respirație și febră în ultimele trei zile. Asociat cu cianoză, pacientul avea antecedente de supt slab și crize de sufocare recurente la fiecare hrănire de la naștere, dar din păcate a fost neglijat. Nu există antecedente de vărsături sau erupții cutanate și nici antecedente de mișcări anormale. Nu are antecedente familiale de avorturi sau decese.
La examinare avea un aspect palid, cașectic și dismorfic (figura 1). (piele hipopegmentată, ochi și păr blond, frunte lată, hiperbolism,filtrum lung și micrognatie). Greutatea sa era de 4.100 g (centila 3-10), lungimea de 53 cm (centila 3-10), iar circumferința capului de 33 cm (centila 3). La examenele oftalmologice a prezentat nistagmus orizontal ușor, cataractă bilaterală și retina flair. Plin de crepitații pe torace bilateral, fără suflu cu zgomot cardiac normal și avea hipotonie profundă.
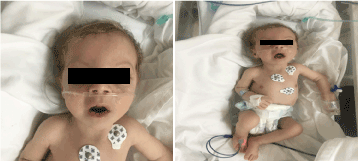
Figura 1. Fotografia clinică a pacientului la vârsta de 2 luni (a) Hipotonie, (b) blond de păr filtrum lung și micrognatie
Investigațiile de laborator au evidențiat anemie microcitară și hipocromică, profilul renal și coagularea au fost neremarcabile. Alanin-transaminaza (ALT) și aspartat aminotransferaza (AST) au fost crescute la 183-231 (10-45 U/L), respectiv 193-301 (10-45 U/L), respectiv 193-301 (10-45 U/L). Bilirubina a fost normală. CK: 389 U/L și LDH: 612 U/L. Amoniacul a fost normal 13 mmol/L. Bilanțul imunologic a evidențiat niveluri reduse de imunoglobuline IgA: 0,1 g/L (0,7-4), dar IgG: 4,51 g/L (7-16), IgM: ,8 g/L (0,4-2,3). S-au încadrat în valoarea normală. Numărul subseturilor de limfocite (CD3, CD4, CD8, CD19 și CD22) a fost normal. Sângele, urina și nu prezentau creșteri. Testul serologic pentru toxoplasmă, rubeolă, citomegalovirus, herpes simplex și markerii hepatici au fost toți normali. Radiografia toracică a evidențiat infiltrație pneumonică și cardiomegalie. Ecografia abdominală a evidențiat hidronefroză bilaterală. Hipertrofia ventriculară stângă a fost detectată prin studiu ecocardiografic. Imagistica prin rezonanță magnetică (IRM) a creierului a confirmat agenezia corpului calos (figura 2). Testele genetice au confirmat o variantă homozigotă de pierdere de funcție a EPG5 (EPG5:NM_020964:exon9:c.1924C>T:p.R642X)
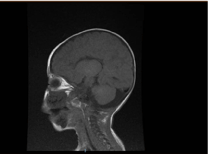
Figura 2. RMN al creierului, secțiune sagitală ponderată T1 care arată agenezie de corp calos
În timpul celei de-a 2-a săptămâni de la internare, starea sa s-a deteriorat rapid cu episoade repetate de pneumonie prin aspirație care au necesitat ventilație. În cele din urmă, pacientul a intrat în stop cardio-respirator și a decedat.
Materiale și metode
WES se efectuează pe ADN genomic utilizând fluxul de lucru Agilent Sure Select Target Enrichment pentru a captura regiunile de interes dintr-o bibliotecă de fragmente de ADN. Întregul exom este secvențiat pe sistemul de secvențiere Illumina HiSeq 2500 cu o acoperire minimă de 30X de 95% din regiunile țintă. Secvențele de ADN din exomul probandului sunt cartografiate și comparate cu secvența de referință hg19 a UCSC hg19, construită pe baza genomului uman. Saudi Diagnostic Laboratories utilizează un pipeline intern pentru a compara secvența probandului cu secvența de referință. Se realizează o evaluare a acoperirii și a calității pentru exonii codificatori vizați din genele RefSeq cunoscute care codifică proteine. Analizele exomului interoghează mii de variante genetice la un proband folosind baze de date proprietare personalizate pentru populațiile arabe. Subansambluri ale acestora sunt caracterizate utilizând liniile directoare ale Colegiului American de Genetică și Genomică Medicală (ACMG) (Richards, et al. 2015) pentru a le clasifica relevanța clinică. Secvențierea Sanger este efectuată pentru a valida toate variantele incluse în raport, cu excepția cazului în care se specifică altfel
Rezultate
La acest pacient a fost identificată o variantă homozigotă de pierdere a funcției EPG5 (EPG5:NM_020964:exon9:c.1924C>T:p.R642X). Conform ghidurilor ACMG (Gen in Med 17:5, 2015), aceasta ar trebui să fie clasificată ca variantă patogenă. Variantele homozigote de pierdere de funcție (LOF) ale EPG5 sunt un mecanism comun al sindromului Vici, 242840 .
Discuție
Sindromul Vici, este o tulburare multi-sistemică congenitală rară care se moștenește într-un model autosomal recesiv, se caracterizează prin insuficiență de creștere, hipopigmentare, defecte imunologice evidente prin infecții recurente, anomalii neurologice de întârziere a dezvoltării, convulsii, cataractă, cardiomiopatie și anomalii neuromusculare; acestea din urmă fiind descrise doar în câteva rapoarte de caz recente . Sindromul Vici a fost raportat pentru prima dată în 1988 de Dionisi Vici, et al. în cazul a doi frați care prezentau albinism, ACC, cataractă, cardiomiopatie, retard mintal și imunodeficiență, iar ulterior a fost denumit „sindromul Vici” de del Campo, et al. în 1999. Sindromul Vici a fost observat atât la bărbați, cât și la femei, și au fost identificate o serie de mutații înrudite în gena EPG5 .
Părinții pacientului nostru au consangvinitate și susțin moștenirea autosomală recesivă a sindromului Vici. Treisprezece dintre cei 31 de pacienți, inclusiv al nostru, sunt frați. Au fost raportate cazuri în care părinții erau veri îndepărtați .
Sindromul Vici se datorează unor mutații recesive în EPG5 pe cromozomul 18q12.3, care codifică proteina 5 a granulelor P ectopice (EPG5), un regulator cheie al autofagiei în organismele superioare. Autofagia este o cale de degradare celulară fundamentală conservată de-a lungul evoluției, cu roluri importante în eliminarea proteinelor și organitelor defecte, apărarea împotriva infecțiilor și adaptarea la cerințele metabolice în schimbare .
Dezvoltarea SNC, reglarea imunitară și pigmentarea pielii la pacienții cu sindromul Vici implică autofagia într-o gamă mai largă de procese celulare. La nivelul creierului, autofagia a fost investigată pe scară largă ca un mecanism patogenic cheie în diverse tulburări neurodegenerative .
Majoritatea semnelor și simptomelor sindromului Vici sunt prezente la naștere; unele caracteristici, cum ar fi cataracta, cardiomiopatia și imunodeficiența, nu sunt întotdeauna prezente la naștere, dar este de așteptat să evolueze în primii ani . Iar cei mai mulți dintre pacienți au decedat în primii trei ani de viață secundar cardiomiopatiei sau pneumoniei
Recunoștințe
Autorii doresc să mulțumească părinților pacientului și laboratorului de diagnostic saudit.
- Dionisi Vici C, Sabetta G, Gambarara M, Vigevano F, Bertini E, et al. (1988) Agenezie de corp calos, imunodeficiență combinată, cataractă bilaterală și hipopigmentare la doi frați. Am J Med Genet 29:1-8.
- Thomas Cullup, Ay Lin Kho, Carlo Dionisi-Vici (2013) Mutațiile recesive în EPG5 cauzează sindromul Vici, o tulburare multisistemică cu autofagie defectuoasă. Nature Genetics 45: 83-87.
- Said E, Soler D, Sewry C (2012) Sindromul Vici – o tulburare neurodegenerativă rapid progresivă cu hipopigmentare, imunodeficiență și modificări miopatice la biopsia musculară. Am J Med Genet A 158: 440-444.
- del Campo M, Hall BD, Aeby A, Nassogne MC, Verloes A, et al. (1999) Albinism și agenezie a corpului calos cu întârziere profundă a dezvoltării: Sindromul Vici, dovezi pentru moștenirea autozomal recesivă. Am J Med Genet 85: 479-485.
- Ehmke N, Parvaneh N, Krawitz P, Ashrafi MR, Karimi P, et al. (2014) Prima descriere a unui pacient cu sindrom Vici din cauza unei mutații care afectează penultimul exon al EPG5 și revizuirea literaturii. Am J Med Genet A 164A: 3170-3175.
- Al-Owain M, Al-Hashem A, Al-Muhaizea M, Humaidan H, Al-Hindi H, et al. (2010) Sindromul Vici asociat cu hipoplazie pulmonară unilaterală și miopatie. Am J Med Genet A 152A: 1849-1853.
- Byrne S, Dionisi-Vici C, Smith L, Gautel M, Jungbluth H, et al. (2016) Sindromul Vici: o revizuire. Orphanet J Rare Dis 11: 21.
- Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, et al. (2006) Proteinele predispuse la agregate sunt eliminate din citosol prin autofagie: implicații terapeutice. Curr Top Dev Biol 76: 89-101.
- Cullup T, Dionisi-Vici C, Kho AL, Yau S, Mohammed S, et al. (2014) Clinical utility gene card for: Sindromul Vici. Eur J Hum Genet 22.
- Curtis Rogers R, Bridgette Aufmuth, Stephanie Monesson (2011) Sindromul Vici: A Rare Autosomal Recessive Syndrome with Brain Anomalies, Cardiomyopathy, and Severe Intellectual Disability (Un sindrom rar autosomal recesiv cu anomalii cerebrale, cardiomiopatie și dizabilitate intelectuală severă). Rapoarte de caz în genetică 2011: 421582.