Odkrywanie i rozwój bezpośrednich inhibitorów czynnika Xa
Czynnik Xa: Structure and binding sitesEdit

Faktory IIa, Xa, VIIa, IXa i XIa są enzymami proteolitycznymi, które pełnią specyficzną rolę w kaskadzie krzepnięcia. Czynnik Xa (FXa) jest najbardziej obiecującym z nich ze względu na jego pozycję na przecięciu szlaku wewnątrzpochodnego i zewnątrzpochodnego, jak również generowanie około 1000 cząsteczek trombiny na każdą cząsteczkę Xa, co skutkuje silnym działaniem przeciwzakrzepowym. FXa powstaje z FX poprzez rozszczepienie 52 aminokwasowego peptydu aktywującego, ponieważ „a” w nazwie czynnika Xa oznacza aktywowany. FXa składa się z 254 aminokwasowej domeny katalitycznej i jest również połączona z 142 aminokwasowym łańcuchem lekkim. Łańcuch ten zawiera zarówno domenę GLA, jak i dwie domeny naskórkowego czynnika wzrostu (EGF like domains).
Miejsce aktywne FXa ma strukturę umożliwiającą katalizowanie rozszczepiania substratów fizjologicznych i rozszczepia PhePheAsnProArg-ThrPhe i TyrIleAspGlyArg-IleVal w protrombinie. FXa posiada cztery tzw. kieszenie, które są celami dla substratów wiążących się z czynnikiem Xa. Kieszenie te wyłożone są różnymi aminokwasami, a inhibitory czynnika Xa wiążąc się z czynnikiem Xa celują właśnie w te kieszenie. Dwie najbardziej istotne kieszenie pod względem powinowactwa i selektywności dla inhibitorów czynnika Xa to S1 i S4.
S1: Kieszeń S1 jest kieszenią hydrofobową i zawiera resztę kwasu asparaginowego (Asp-189), która może służyć jako miejsce rozpoznania dla grupy zasadowej. FXa posiada w kieszeni S1 przestrzeń resztową, która jest wyścielona resztami Tyr-228, Asp-189 i Ser-195.
S2: Kieszeń S2 jest małą i płytką kieszenią. Łączy się z kieszenią S4 i ma miejsce dla małych aminokwasów. Tyr-99 wydaje się blokować dostęp do tej kieszeni, więc ta kieszeń nie jest tak ważna jak S1 i S4.
S3: Kieszeń S3 znajduje się na obręczy kieszeni S1, jest płaska i wystawiona na działanie rozpuszczalnika. Kieszeń ta nie jest tak istotna jak S1 i S4.
S4: Kieszeń S4 ma charakter hydrofobowy, a dno kieszeni tworzy reszta Trp-215. Reszty Phe-174 i Tyr-99 FXa łączą się z Trp-215 tworząc aromatyczne pudełko, które jest zdolne do wiązania fragmentów alifatycznych, aromatycznych i dodatnio naładowanych. Ze względu na wiązanie z dodatnio naładowanymi jednostkami, można ją określić jako dziurę kationową.
Struktura chemiczna i właściwości bezpośrednich inhibitorów XaEdit
| Rivaroxaban | Apixaban | Edoxaban | ||
|---|---|---|---|---|
| MW (g/mol) | 436 | 460 | 548 | |
| Wzór cząsteczkowy wzór | C19H18ClN3O5S | C25H25N5O4 | C24H30ClN7O4S | |
| Kształt | L | L | L | |
| Ki | 0.4 nM | 0,08 nM | 0,561 nM | |
| IC50 | 0.7 nM | N/A | N/A | |
| Dostępność biologiczna w jamie ustnej (%) | 66-100 (zależna od dawki) | 66-100 (zależna od dawki)zależna od dawki) | 50 | 62 |

Wiązanie inhibitorów czynnika Xa z czynnikiem XaEdit
Inhibitory czynnika Xa wszystkie wiążą się w tzw. sposób L-shape w obrębie miejsca aktywnego czynnika Xa. Kluczowymi składnikami czynnika Xa są miejsca wiążące S1 i S4. Po raz pierwszy zauważono, że naturalne związki, antistasin i TAP, które posiadają wysoce polarne, a więc naładowane składniki, wiążą się z celem z pewną specyficznością. Dlatego zaprojektowano nowsze leki z dodatnio naładowanymi grupami, ale skutkowały one słabą biodostępnością. Obecnie dostępne na rynku inhibitory czynnika Xa zawierają zatem pierścień aromatyczny z różnymi cząsteczkami przyłączonymi w celu uzyskania różnych interakcji z miejscami wiązania S1 i S4. Zapewnia to również dobrą biodostępność, jak również utrzymanie siły wiązania. Inhibitory czynnika Xa dostępne obecnie na rynku opierają się zatem na wiązaniach hydrofobowych i wodorowych zamiast na wysoce polarnych interakcjach.
Antystasyna wiążąca się z czynnikiem XaEdit
Antystasyna zawiera domenę N- i C-końcową, które są podobne w swoich sekwencjach aminokwasowych z ~40% identycznością i ~56% homologią. Każda z nich zawiera krótką strukturę arkusza β i 5 wiązań disulfidowych. Tylko domena N-końcowa jest niezbędna do hamowania Xa, podczas gdy domena C-końcowa nie przyczynia się do właściwości inhibicyjnych ze względu na różnice w strukturze trójwymiarowej, mimo że domena C-końcowa ma silnie analogiczny wzór do rzeczywistego miejsca aktywnego.
Interakcja antystazyny z FXa obejmuje zarówno miejsce aktywne, jak i nieaktywną powierzchnię FXa. Miejsce reaktywne antystaszyny utworzone przez Arg-34 i Val-35 w N-końcowej domenie odpowiada miejscu wiązania FXa, najprawdopodobniej kieszeni S1. Jednocześnie, Glu-15 znajdujący się poza miejscem reaktywnym antystaszyny pasuje do dodatnio naładowanych reszt na powierzchni FXa. Wiązanie wielokrotne jest termodynamicznie korzystne i prowadzi do sub-nanomolarnej inhibicji (Ki = 0,3-0,6 nM).
Wiązanie DX-9065a z czynnikiem XaEdit
DX-9065a, pierwszy małocząsteczkowy bezpośredni inhibitor Xa, jest pochodną amidinoarylową o masie cząsteczkowej 571,07g/mol. Jego dodatnio naładowana grupa amidinonaftalenowa tworzy mostek solny do reszty Asp-189 w kieszeni S1 FXa. Pierścień pirrolidynowy mieści się pomiędzy Tyr-99, Phe-174 i Trp-215 w kieszeni S4 FXa.
W przeciwieństwie do starszych leków, np. heparyny, DX-9065a jest selektywny dla FXa w porównaniu z trombiną, mimo że FXa i trombina mają podobną budowę. Jest to spowodowane różnicą w resztach aminokwasowych w pozycji homologicznej 192. Podczas gdy FXa ma w tej pozycji resztę glutaminową, trombina ma kwas glutaminowy, który powoduje odpychanie elektrostatyczne z grupą karboksylową DX-9065a. Ponadto mostek solny pomiędzy Glu-97 trombiny a grupą amidynową umocowaną w pierścieniu pirolidynowym DX-9065a zmniejsza elastyczność cząsteczki DX-9065a, która nie może teraz obrócić się na tyle, aby uniknąć zderzenia elektrostatycznego. Dlatego wartość IC50 dla trombiny wynosi >1000µM, podczas gdy wartość IC50 dla FXa wynosi 0,16µM.

Wiązanie rywaroksabanu z czynnikiem XaEdit
Wiązanie rywaroksabanu z FXa odbywa się za pośrednictwem dwóch wiązań wodorowych z aminokwasem Gly-219. Te dwa wiązania wodorowe pełnią ważną rolę w kierowaniu leku do miejsc S1 i S4 FXa. Pierwsze wiązanie wodorowe jest silnym oddziaływaniem, które pochodzi od tlenu karbonylowego rdzenia oksazolidynonowego rywaroksabanu. Drugie wiązanie wodorowe jest słabszą interakcją i pochodzi od grupy aminowej karboksyamidowej cząsteczki klorotiofenowej.
Te dwa wiązania wodorowe powodują, że lek tworzy kształt litery L i mieści się w kieszeniach S1 i S4. Reszty aminokwasowe Phe-174, Tyr-99 i Trp-215 tworzą wąski kanał hydrofobowy, który jest kieszenią wiążącą S4. Część morfolinonowa rywaroksabanu jest „wciśnięta” pomiędzy aminokwasy Tyr-99 i Phe-174, a pierścień arylowy rywaroksabanu jest zorientowany prostopadle do Trp-215. Grupa karbonylowa morfolinonu nie ma bezpośredniej interakcji ze szkieletem FXa, zamiast tego przyczynia się do planaryzacji pierścienia morfolinonu i dlatego wspiera riwaroksaban w umieszczeniu między dwoma aminokwasami.
Interakcja między podstawnikiem chlorowym cząsteczki tiofenowej a pierścieniem aromatycznym Tyr-228, który znajduje się na dole S1, jest bardzo ważna ze względu na fakt, że eliminuje potrzebę silnie zasadowych grup dla wysokiego powinowactwa do FXa. Dzięki temu rywaroksaban, który jest niebazowy, może osiągnąć dobrą doustną biodostępność i siłę działania.

Wiązanie apiksabanu z czynnikiem XaEdit
Apiksaban wykazuje podobny sposób wiązania jak rywaroksaban i tworzy ścisły kompleks inhibitor-enzym po przyłączeniu do FXa. Grupa p-metoksy apiksabanu łączy się z kieszenią S1 FXa, ale nie wydaje się mieć żadnych interakcji z żadnymi resztami w tym regionie FXa. Atom azotu pirazolu N-2 apiksabanu oddziałuje z Gln-192, a tlen karbonylowy oddziałuje z Gly-216. Grupa fenylo-laktamowa apiksabanu znajduje się pomiędzy Tyr-99 a Phe-174 i dzięki swojej orientacji może oddziaływać z Trp-215 kieszeni S4. Karbonylowa grupa tlenowa cząsteczki laktamowej oddziałuje z cząsteczką wody i nie wydaje się oddziaływać z żadnymi resztami w kieszeni S4.

Zależność struktura-aktywność-relacja (SAR)Edycja
Ważną częścią projektowania związku, który jest idealnym inhibitorem określonego celu, jest zrozumienie sekwencji aminokwasowej miejsca docelowego, do którego związek ma się wiązać. Modelowanie zarówno protrombiny jak i FXa pozwala na odjęcie różnic i zidentyfikowanie aminokwasów w każdym miejscu wiązania. Na dnie kieszeni S1 w FXa aminokwasem wiążącym jest Asp-189, z którym mogą wiązać się cząsteczki amidynowe. Po prześwietleniu miejsca wiązania FXa okazało się, że kieszeń S1 ma planarny kształt, co oznacza, że płaska grupa amidinoarylowa powinna się do niej wiązać bez przeszkód sterycznych.
Nowoczesne bezpośrednie inhibitory Xa to cząsteczki w kształcie litery L, których końce idealnie pasują do kieszeni S1 i S4. Długa strona kształtu L musi być zgodna z wysoce specyficznym tunelem w miejscu aktywnym celu. Aby to osiągnąć, ta część cząsteczki została zaprojektowana tak, aby miała niewiele formalnych interakcji z FXa w tym regionie. Ponieważ nie ma tam specyficznego wiązania, dopasowanie tych czynników pomiędzy kieszeniami FXa zwiększa całkowitą specyficzność leków w stosunku do cząsteczki FXa. Interakcja między kieszenią S1 FXa a inhibitorem może być zarówno jonowa, jak i niejonowa, co jest ważne, ponieważ pozwala na dostosowanie konstrukcji cząsteczki w celu zwiększenia doustnej biodostępności. Wcześniej zaprojektowane związki były cząsteczkami naładowanymi, które nie wchłaniają się dobrze w przewodzie pokarmowym i dlatego nie osiągały wysokich stężeń w surowicy. Nowsze leki mają lepszą biodostępność, ponieważ nie są naładowane i mają niejonową interakcję z kieszenią S1.
Rivaroxaban
Podczas opracowywania SAR dla rivaroxabanu badacze zdali sobie sprawę, że dodanie grupy 5-chlorotiofen-2-karboksyamidowej do rdzenia oksazolidoniny może zwiększyć siłę działania o 200 razy, która wcześniej była zbyt słaba do zastosowań medycznych. Oprócz tego odkrycia potwierdzono wyraźną preferencję dla konfiguracji (S)-. Związek ten miał obiecujący profil farmakokinetyczny i nie zawierał wysoce zasadowej grupy amidynowej, która wcześniej była uważana za istotną dla interakcji z kieszenią S1. Wyniki te doprowadziły do szeroko zakrojonych badań SAR (structure-activity relationship). Podczas badań SAR, R1 została zdefiniowana jako najważniejsza grupa dla siły działania. Pierwszą grupą funkcyjną R1, która znacząco zwiększyła siłę działania był pirolidynon, ale dalsze badania wykazały jeszcze większą siłę działania z grupą morfolinonową. Grupy R2 i R3 miały dołączony wodór lub fluor i szybko oceniono, że posiadanie wodoru spowodowało najwyższą siłę działania. Grupy R2 i R3 zostały następnie zastąpione różnymi grupami, z których wszystkie były mniej silne niż wodór, więc wodór był ostatecznym wynikiem. Ponieważ część cząsteczki chlorotiofenowej miała nieodpowiednią rozpuszczalność w wodzie, próbowano zastąpić ją inną grupą, ale nie powiodło się. Cząsteczka chlorotiofenowa wiąże się z Tyr-228 w dolnej części kieszeni S1, co czyni ją kluczowym czynnikiem w wiązaniu z FXa. Rivaroxaban ma zarówno wysokie powinowactwo, jak i dobrą biodostępność.

Apiksaban

Podczas opracowywania SAR apiksabanu istniały trzy grupy, które należało przetestować w celu uzyskania maksymalnej siły działania i biodostępności. Pierwszą grupą do zbadania było miejsce nieaktywne, ponieważ musi być ono ustabilizowane przed badaniem SAR na grupie p-metoksyfenylowej (cząsteczka wiążąca S1). Istnieje kilka grup, które zwiększają siłę działania związku, głównie amidy, aminy i tetrazole, ale także grupy metylosulfonylowe i trifluorometylowe. Z tych grup karboksyamid ma największe wiązanie i miał podobną aktywność krzepnięcia jak związki.
W badaniach na psach, ten związek z grupą karboksyamidową o nazwie 13F, wykazywał świetny profil farmakokinetyczny, niski klirens i odpowiedni okres półtrwania i objętość dystrybucji. Ze względu na sukces w znalezieniu grupy stabilizującej zaprzestano badań SAR dla grupy wiążącej S1 (p-metoksyfenylu). W grupie wiążącej S4 analogi N-metyloacetylowe i laktamowe wykazały bardzo wysokie powinowactwo wiązania do FXa, dużą krzepliwość i selektywność w stosunku do innych proteaz. Istotna okazała się orientacja, gdyż N-metyloacetyl, w porównaniu z acetamidem, miał 300-krotnie mniejszą zdolność wiązania z FXa ze względu na niekorzystną planaryzację w pobliżu miejsca wiązania regionu S4.
SynthesisEdit
RivaroxabanEdit
Rivaroxaban chemicznie należy do grupy n-aryloxazolidinonów. Inne leki z tej grupy to linezolid i tedizolid, z których oba są antybiotykami. Synteza n-aryloksazolidynonów rozpoczynająca się od chronionego O-silylem karbaminianu etylu(2,3-dihydroksypropylu)została opublikowana w 2016 roku. W reakcji typu one-pot karbaminian ulega cyklizacji do pierścienia 2-oksazolidonowego w warunkach lekko zasadowych, podczas gdy jednocześnie azot oksazolidonowy ulega aryzacji w wyniku katalizy miedziowej. Szczególnie w przypadku rywaroksabanu, 3-morfolinon zastępuje jod w pozycji p pierścienia benzenowego w procesie katalizy miedziowej. Następnie usuwa się sililową grupę ochronną, a powstały alkohol zastępuje się grupą aminową, która w ostatnim etapie jest acylowana.
Przemysłowy preparat rivaroxabanu został zarejestrowany jako patent przez Bayer Healthcare w 2005 roku. Rozpoczyna się on od N-(4-aminofenol)-morfolinonu, który jest alkilowany przez pochodną tlenku propylenu, zawierającą również aminę pierwszorzędową związaną z ftalimidową grupą ochronną. Następnie dodaje się odpowiednik fosgenu w celu utworzenia pierścienia 2-oksazolidonowego i usuwa się ftalimid. Wolna amina może być teraz acylowana, co prowadzi do powstania rywaroksabanu.
Jednakże według patentu synteza ta ma „różne wady w zarządzaniu reakcją, co ma szczególnie niekorzystne skutki dla preparatu”. Patent wyjaśnia również inną syntezę rozpoczynającą się od pochodnej chlorotiofenu, która byłaby bardziej odpowiednia dla procesu przemysłowego, ale zwraca uwagę, że toksyczne rozpuszczalniki lub odczynniki muszą być usunięte z produktu końcowego. Dlatego sposób ten nie stanowi alternatywy.
Opisano różne inne ścieżki syntezy rywaroksabanu.
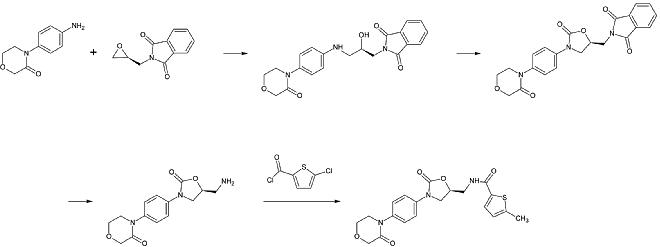
1. etap: Alkilowanie pierwszorzędowej aminy aromatycznej
2. krok: Tworzenie pierścienia 2-oksazolinidonu, przy użyciu odpowiednika fosgenu
3. krok: Usunięcie ftalimidowej grupy ochronnej
4. krok: Acylowanie pierwszorzędowej aminy
ApiksabanEdit
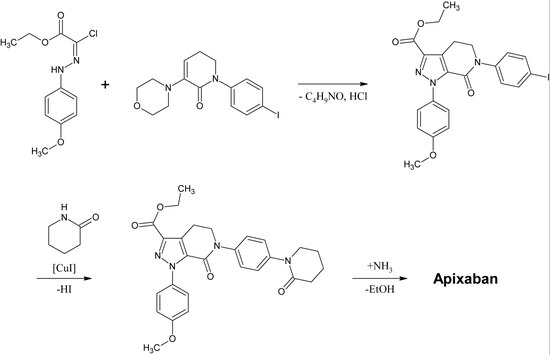
Pierwsza pełna synteza apiksabanu została opublikowana w 2007 roku. Kluczowym etapem tej reakcji jest (3+2)cykloaddycja pochodnej p-metoksyfenylochlorohydrazonu i pochodnej p-jodofenylo-morfoliny-dihydropirydyny. Po następującej eliminacji HCl i morfoliny, jod podstawia się 2-piperydynonem na drodze katalizy miedziowej, a ester etylowy przekształca się w amid (aminoliza). Reakcja ta została zarejestrowana jako patent w 2009 roku.
.