Entdeckung und Entwicklung von direkten Xa-Inhibitoren
Faktor Xa: Struktur und BindungsstellenEdit

Die Faktoren IIa, Xa, VIIa, IXa und XIa sind allesamt proteolytische Enzyme, die eine spezifische Rolle in der Gerinnungskaskade spielen. Faktor Xa (FXa) ist das vielversprechendste Enzym, da es an der Schnittstelle zwischen dem intrinsischen und dem extrinsischen Weg liegt und für jedes Xa-Molekül etwa 1000 Thrombinmoleküle erzeugt, was zu einer starken gerinnungshemmenden Wirkung führt. FXa wird aus FX durch Spaltung eines 52 Aminosäuren umfassenden Aktivierungspeptids gebildet, da das „a“ in Faktor Xa für aktiviert steht. FXa besteht aus einer katalytischen Domäne mit 254 Aminosäuren und ist außerdem mit einer leichten Kette mit 142 Aminosäuren verbunden. Die Kette enthält sowohl die GLA-Domäne als auch zwei Domänen des epidermalen Wachstumsfaktors (EGF-ähnliche Domänen).
Das aktive Zentrum von FXa ist so strukturiert, dass es die Spaltung physiologischer Substrate katalysiert und PhePheAsnProArg-ThrPhe und TyrIleAspGlyArg-IleVal in Prothrombin spaltet. FXa verfügt über vier so genannte Taschen, die als Ziel für die Bindung von Substraten an Faktor Xa dienen. Diese Taschen werden von verschiedenen Aminosäuren ausgekleidet, und Xa-Inhibitoren zielen bei der Bindung an Faktor Xa auf diese Taschen ab. Die beiden für die Affinität und Selektivität der Xa-Inhibitoren wichtigsten Taschen sind S1 und S4.
S1: Die S1-Tasche ist eine hydrophobe Tasche und enthält einen Asparaginsäurerest (Asp-189), der als Erkennungsstelle für eine basische Gruppe dienen kann. FXa hat einen Restraum in der S1-Tasche, der von den Resten Tyr-228, Asp-189 und Ser-195 ausgekleidet ist.
S2: Die S2-Tasche ist eine kleine und flache Tasche. Sie geht in die S4-Tasche über und bietet Platz für kleine Aminosäuren. Tyr-99 scheint den Zugang zu dieser Tasche zu blockieren, so dass diese Tasche nicht so wichtig ist wie S1 und S4.
S3: Die S3-Tasche befindet sich am Rande der S1-Tasche, ist flach und dem Lösungsmittel ausgesetzt. Diese Tasche ist nicht so wichtig wie S1 und S4.
S4: Die S4-Tasche ist von Natur aus hydrophob und der Boden der Tasche wird vom Trp-215-Rest gebildet. Die Reste Phe-174 und Tyr-99 von FXa bilden zusammen mit Trp-215 eine aromatische Box, die in der Lage ist, aliphatische, aromatische und positiv geladene Fragmente zu binden. Aufgrund der Bindung an positiv geladene Einheiten kann es als Kationenloch bezeichnet werden.
Chemische Struktur und Eigenschaften von direkten Xa-InhibitorenBearbeiten
| Rivaroxaban | Apixaban | Edoxaban | |
|---|---|---|---|
| MW (g/mol) | 436 | 460 | 548 |
| Mol. Formel | C19H18ClN3O5S | C25H25N5O4 | C24H30ClN7O4S |
| Form | L | L | L |
| Ki | 0.4 nM | 0,08 nM | 0.561 nM |
| IC50 | 0.7 nM | N/A | N/A |
| Orale Bioverfügbarkeit (%) | 66-100 (dosis-abhängig) | 50 | 62 |

Bindung von Xa-Inhibitoren an Faktor XaEdit
Die Xa-Inhibitoren binden alle in einer so genannten L-Form im aktiven Zentrum von Faktor Xa. Die wichtigsten Bestandteile des Faktor Xa sind die Bindungsstellen S1 und S4. Es wurde zunächst festgestellt, dass die natürlichen Verbindungen Antistasin und TAP, die stark polare und daher geladene Komponenten besitzen, mit einer gewissen Spezifität an das Ziel binden. Aus diesem Grund wurden neuere Medikamente mit positiv geladenen Gruppen entwickelt, die jedoch eine schlechte Bioverfügbarkeit aufwiesen. Die heute auf dem Markt befindlichen Xa-Inhibitoren enthalten daher einen aromatischen Ring mit verschiedenen Komponenten, die für unterschiedliche Wechselwirkungen mit den S1- und S4-Bindungsstellen sorgen. Dies gewährleistet ebenfalls eine gute Bioverfügbarkeit und eine feste Bindungsstärke. Die heute auf dem Markt befindlichen Xa-Inhibitoren beruhen daher auf hydrophoben und Wasserstoffbrückenbindungen anstelle von hochpolaren Wechselwirkungen.
Antistasin bindet an Faktor XaEdit
Antistasin enthält eine N- und eine C-terminale Domäne, die sich in ihren Aminosäuresequenzen mit ~40% Identität und ~56% Homologie ähneln. Jede von ihnen enthält eine kurze β-Faltblattstruktur und 5 Disulfidbindungen. Nur die N-terminale Domäne ist notwendig, um Xa zu hemmen, während die C-terminale Domäne aufgrund von Unterschieden in der dreidimensionalen Struktur nicht zu den hemmenden Eigenschaften beiträgt, obwohl die C-terminale Domäne ein stark analoges Muster zur eigentlichen aktiven Stelle aufweist.
Die Wechselwirkung von Antistasin mit FXa betrifft sowohl die aktive Stelle als auch die inaktive Oberfläche von FXa. Die reaktive Stelle von Antistasin, die von Arg-34 und Val-35 in der N-terminalen Domäne gebildet wird, passt zur Bindungsstelle von FXa, höchstwahrscheinlich die S1-Tasche. Gleichzeitig passt Glu-15, das sich außerhalb der reaktiven Stelle von Antistasin befindet, zu positiv geladenen Resten auf der Oberfläche von FXa. Die Mehrfachbindung ist thermodynamisch vorteilhaft und führt zu einer subnanomolaren Hemmung (Ki = 0,3-0,6 nM).
DX-9065a bindet an Faktor XaEdit
DX-9065a, der erste niedermolekulare direkte Xa-Inhibitor, ist ein Amidinoaryl-Derivat mit einem Molekulargewicht von 571,07g/mol. Seine positiv geladene Amidinonaphtalene-Gruppe bildet eine Salzbrücke zum Asp-189-Rest in der S1-Tasche von FXa. Der Pyrrolidinring sitzt zwischen Tyr-99, Phe-174 und Trp-215 in der S4-Tasche von FXa.
Im Gegensatz zu älteren Medikamenten, z.B. Heparin, ist DX-9065a selektiv für FXa im Vergleich zu Thrombin, obwohl FXa und Thrombin in ihrer Struktur ähnlich sind. Dies wird durch einen Unterschied im Aminosäurerest in der homologen Position 192 verursacht. Während FXa an dieser Stelle einen Glutaminrest aufweist, hat Thrombin eine Glutaminsäure, die eine elektrostatische Abstoßung mit der Carboxylgruppe von DX-9065a verursacht. Darüber hinaus verringert eine Salzbrücke zwischen Glu-97 von Thrombin und der im Pyrrolidinring von DX-9065a fixierten Amidingruppe die Flexibilität des DX-9065a-Moleküls, das sich nun nicht mehr ausreichend drehen kann, um den elektrostatischen Zusammenstoß zu vermeiden. Deshalb beträgt der IC50-Wert für Thrombin >1000µM, während der IC50-Wert für FXa 0,16µM beträgt.

Rivaroxaban bindet an Faktor XaEdit
Die Bindung von Rivaroxaban an FXa wird durch zwei Wasserstoffbrückenbindungen zur Aminosäure Gly-219 vermittelt. Diese beiden Wasserstoffbrücken spielen eine wichtige Rolle bei der Bindung des Medikaments an die S1- und S4-Subsites von FXa. Die erste Wasserstoffbrückenbindung ist eine starke Wechselwirkung, die vom Carbonylsauerstoff des Oxazolidinonkerns von Rivaroxaban ausgeht. Bei der zweiten Wasserstoffbrücke handelt es sich um eine schwächere Wechselwirkung, die von der Aminogruppe des Clorothiophen-Carboxamid-Teils ausgeht.
Diese beiden Wasserstoffbrücken führen dazu, dass das Medikament eine L-Form bildet und in die S1- und S4-Taschen passt. Die Aminosäurereste Phe-174, Tyr-99 und Trp-215 bilden einen engen hydrophoben Kanal, der die S4-Bindungstasche darstellt. Der Morpholinonteil von Rivaroxaban ist zwischen den Aminosäuren Tyr-99 und Phe-174 „eingeklemmt“, und der Arylring von Rivaroxaban ist senkrecht über Trp-215 ausgerichtet. Die Morpholinon-Carbonylgruppe hat keine direkte Wechselwirkung mit dem FXa-Rückgrat, sondern trägt zu einer Planarisierung des Morpholinonrings bei und unterstützt daher die Einbettung von Rivaroxaban zwischen den beiden Aminosäuren.
Die Wechselwirkung zwischen dem Chlorsubstituenten des Thiophenanteils und dem aromatischen Ring von Tyr-228, der sich am unteren Ende von S1 befindet, ist sehr wichtig, da sie die Notwendigkeit stark basischer Gruppen für eine hohe Affinität zu FXa überflüssig macht. Dadurch kann Rivaroxaban, das nicht basisch ist, eine gute orale Bioverfügbarkeit und Wirksamkeit erreichen.

Apixaban bindet an Faktor XaEdit
Apixaban zeigt einen ähnlichen Bindungsmodus wie Rivaroxaban und bildet einen engen Inhibitor-Enzym-Komplex, wenn es mit FXa verbunden ist. Die p-Methoxygruppe von Apixaban verbindet sich mit der S1-Tasche von FXa, scheint aber keine Wechselwirkung mit irgendwelchen Resten in dieser Region von FXa zu haben. Das N-2-Stickstoffatom des Pyrazols von Apixaban interagiert mit Gln-192 und der Carbonylsauerstoff interagiert mit Gly-216. Die Phenyllactamgruppe von Apixaban befindet sich zwischen Tyr-99 und Phe-174 und ist aufgrund ihrer Ausrichtung in der Lage, mit Trp-215 der S4-Tasche zu interagieren. Die Carbonylsauerstoffgruppe der Lactameinheit interagiert mit einem Wassermolekül und scheint nicht mit irgendwelchen Resten in der S4-Tasche zu interagieren.

Struktur-Aktivitäts-Beziehung (SAR)Bearbeiten
Ein wichtiger Teil des Entwurfs einer Verbindung, die ein idealer Hemmstoff für ein bestimmtes Ziel ist, besteht darin, die Aminosäuresequenz der Zielstelle zu verstehen, an die die Verbindung binden soll. Die Modellierung sowohl von Prothrombin als auch von FXa ermöglicht es, den Unterschied zu erkennen und die Aminosäuren an jeder Bindungsstelle zu identifizieren. Am Boden der S1-Tasche von FXa ist die bindende Aminosäure Asp-189, an die Amidineinheiten binden können. Nach der Röntgenuntersuchung der Bindungsstelle von FXa wurde festgestellt, dass die S1-Tasche eine planare Form hat, was bedeutet, dass eine flache Amidinoarylgruppe ohne sterische Hindernisse daran binden sollte.
Moderne direkte Xa-Inhibitoren sind L-förmige Moleküle, deren Enden perfekt in die S1- und S4-Taschen passen. Die lange Seite des L-förmigen Moleküls muss in einen hochspezifischen Tunnel im aktiven Zentrum des Zielmoleküls passen. Um dies zu erreichen, ist dieser Teil der Moleküle so konzipiert, dass er nur geringe formale Wechselwirkungen mit FXa in dieser Region aufweist. Da es keine spezifische Bindung gibt, erhöht die Passung dieser Wirkstoffe zwischen den Taschen von FXa die Gesamtspezifität der Medikamente für das FXa-Molekül. Die Wechselwirkung zwischen der S1-Tasche von FXa und dem Inhibitor kann sowohl ionisch als auch nicht-ionisch sein, was wichtig ist, weil dadurch das Design der Komponente angepasst werden kann, um die orale Bioverfügbarkeit zu erhöhen. Bei den früher entwickelten Verbindungen handelte es sich um geladene Moleküle, die im Magen-Darm-Trakt nicht gut absorbiert werden und daher keine hohen Serumkonzentrationen erreichen. Die neueren Medikamente haben eine bessere Bioverfügbarkeit, da sie nicht geladen sind und eine nicht-ionische Wechselwirkung mit der S1-Tasche haben.
Rivaroxaban
Während der SAR-Entwicklung von Rivaroxaban erkannten die Forscher, dass das Hinzufügen einer 5-Chlorthiophen-2-carboxamid-Gruppe zum Oxazolidonin-Kern die Potenz um das 200-fache erhöhen könnte, die zuvor für eine medizinische Verwendung zu schwach war. Zusätzlich zu dieser Entdeckung wurde eine klare Präferenz für die (S)-Konfiguration bestätigt. Diese Verbindung wies ein vielversprechendes pharmakokinetisches Profil auf und enthielt keine hochgradig basische Amidingruppe, die jedoch zuvor als wichtig für die Interaktion mit der S1-Tasche angesehen worden war. Diese Erkenntnisse führten zu umfangreichen SAR-Untersuchungen (Struktur-Wirkungs-Beziehung). Bei den SAR-Tests wurde R1 als die für die Wirksamkeit wichtigste Gruppe definiert. Pyrrolidinon war die erste funktionelle R1-Gruppe, die die Potenz signifikant erhöhte, aber weitere Untersuchungen ergaben eine noch höhere Potenz mit einer Morpholinon-Gruppe. An die Gruppen R2 und R3 wurde Wasserstoff oder Fluor angehängt, und es wurde schnell festgestellt, dass Wasserstoff die höchste Potenz ergibt. Die Gruppen R2 und R3 wurden dann durch verschiedene Gruppen ersetzt, die alle weniger wirksam waren als die Wasserstoffgruppe, so dass Wasserstoff das Endergebnis war. Da die Chlorthiophengruppe nur unzureichend wasserlöslich war, wurde versucht, sie durch eine andere Gruppe zu ersetzen, was jedoch nicht gelang. Die Chlorthiophengruppe bindet an Tyr-228 am unteren Ende der S1-Tasche und ist damit ein Schlüsselfaktor für die Bindung an FXa. Rivaroxaban hat sowohl eine hohe Affinität als auch eine gute Bioverfügbarkeit.

Apixaban

Während der SAR-Entwicklung von Apixaban mussten drei Gruppen getestet werden, um eine maximale Wirksamkeit und Bioverfügbarkeit zu erreichen. Die erste Gruppe, die getestet werden musste, war die nicht aktive Stelle, da sie vor dem SAR-Test an der p-Methoxyphenylgruppe (S1-Bindungsteil) stabilisiert werden muss. Es gibt mehrere Gruppen, die die Potenz der Verbindung erhöhen, hauptsächlich Amide, Amine und Tetrazole, aber auch Methylsulfonyl- und Trifluormethylgruppen. Von diesen Gruppen hat die Carboxamidgruppe die stärkste Bindungswirkung und zeigte eine ähnliche Gerinnungsaktivität wie die anderen Verbindungen.
In Tests mit Hunden zeigte diese Verbindung mit einer Carboxamidgruppe, genannt 13F, ein hervorragendes pharmakokinetisches Profil, eine geringe Clearance und eine angemessene Halbwertszeit und ein angemessenes Verteilungsvolumen. Aufgrund des Erfolgs bei der Suche nach einer stabilisierenden Gruppe wurde die SAR-Forschung für die S1-Bindungsgruppe (p-Methoxyphenyl) eingestellt. In der S4-Bindungsgruppe erwiesen sich die N-Methylacetyl- und Lactam-Analoga als sehr bindungsaffin für FXa und zeigten eine hohe Gerinnungsfähigkeit und Selektivität gegenüber anderen Proteasen. Die Orientierung erwies sich als wichtig, da N-Methylacetyl im Vergleich zu Acetamid eine 300-fach geringere Bindungsfähigkeit an FXa aufwies, was auf eine ungünstige Planarität in der Nähe der Bindungsstelle in der S4-Region zurückzuführen ist.
SyntheseBearbeiten
RivaroxabanBearbeiten
Rivaroxaban gehört chemisch gesehen zur Gruppe der n-Aryloxazolidinone. Andere Arzneimittel dieser Gruppe sind Linezolid und Tedizolid, die beide Antibiotika sind. Eine Synthese von n-Aryloxazolidinonen ausgehend von einem O-Silyl-geschützten Ethyl(2,3-dihydroxypropyl)-carbamat wurde 2016 veröffentlicht. In einer Eintopfreaktion cyclisiert das Carbamat unter leicht basischen Bedingungen zu einem 2-Oxazolidonring, während gleichzeitig der Oxazolidonstickstoff durch Kupferkatalyse aryliert wird. Insbesondere bei Rivaroxaban ersetzt 3-Morpholinon das Iod in p-Position des Benzolrings durch Kupfer-Katalyse. Anschließend wird die Silyl-Schutzgruppe entfernt und der resultierende Alkohol durch eine Aminogruppe ersetzt, die dann im letzten Schritt acyliert wird.
Eine industrielle Zubereitung von Rivaroxaban wurde 2005 von Bayer Healthcare zum Patent angemeldet. Sie geht von N-(4-Aminophenol)-morpholinon aus, das mit einem Propylenoxid-Derivat alkyliert wird, das ebenfalls ein primäres Amin enthält, das an einer Phthalimid-Schutzgruppe beteiligt ist. Anschließend wird ein Phosgenäquivalent hinzugefügt, um den 2-Oxazolidonring zu bilden, und das Phthalimid wird entfernt. Das freie Amin kann nun acyliert werden, was zu Rivaroxaban führt.
Die Synthese hat jedoch laut Patent „verschiedene Nachteile in der Reaktionsführung, die sich besonders ungünstig auf die Zubereitung auswirken“. Das Patent erläutert auch eine andere Synthese ausgehend von einem Chlorthiophen-Derivat, die für den industriellen Prozess besser geeignet wäre, weist aber darauf hin, dass giftige Lösungsmittel oder Reagenzien aus dem Endprodukt entfernt werden müssen. Daher ist dieser Weg keine Alternative.
Verschiedene andere Synthesewege von Rivaroxaban wurden beschrieben.
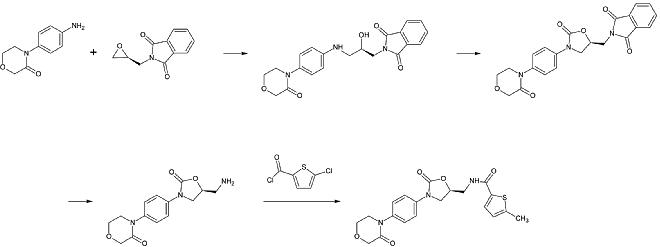
1. Schritt: Alkylierung des primären aromatischen Amins
2. Schritt: Bildung des 2-Oxazolinidon-Rings unter Verwendung eines Phosgen-Äquivalents
3. Schritt: Abspaltung der Phthalimid-Schutzgruppe
4.Schritt: Acylierung des primären Amins
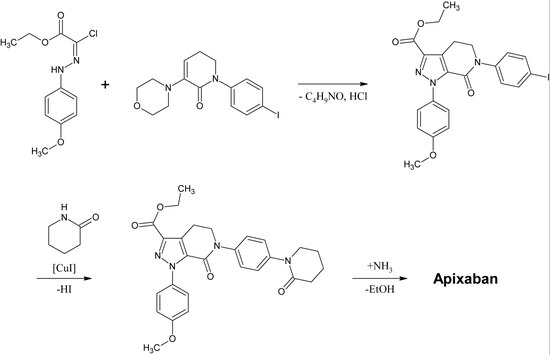
ApixabanEdit
Die erste vollständige Synthese von Apixaban wurde im Jahr 2007 veröffentlicht. Der Schlüsselschritt dieser Reaktion ist eine (3+2)-Cycloaddition eines p-Methoxyphenylchlorhydrazon-Derivats und eines p-Iodophenylmorpholin-Dihydropyridin-Derivats. Nach der anschließenden Abspaltung von HCl und Morpholin wird das Iod durch Kupferkatalyse durch 2-Piperidinon substituiert und der Ethylester in ein Amid umgewandelt (Aminolyse). Diese Reaktion wurde 2009 zum Patent angemeldet.