最近の記事
要旨
目的:重症低血圧、心筋症、体幹無形成を有する乳児に認められた眼皮膚色素沈着の症例を報告すること
事例報告。 妊娠37週で出産した男児を対象とした研究である。 健康なYamani家の両親から生まれ、成長障害、先天性両白内障、顔面・頭髪・皮膚の異形、眼振があり、左心室肥大を有していた。 Vici症候群の原因としてepg5という遺伝子の変異が同定されている
結論。 Vici症候群は明確な臨床像である。 その主な臨床症状は,低月経,発達遅延,白子,心筋症,免疫不全,脳梁の奇形などであった。
キーワード
Vici症候群;脳梁発育不全。 EPG5
はじめに
Vici症候群は、脳梁奇形、白内障、眼皮膚色素沈着、心筋症、成長不全、変型を主要徴とする劣性遺伝性の重症先天異常であり、その原因は、脳梁奇形、眼皮膚色素沈着、心筋症、成長不全、変型にある。
この疾患は1988年に、脳梁奇形、皮膚色素沈着、両側白内障、口唇口蓋裂、複合免疫不全からなる奇形症候群の2人の兄弟で初めて報告されました。 兄弟は重度の精神運動障害、発作、重度の呼吸器感染症の再発、慢性粘膜皮膚カンジダ症に苦しんだ。 Vici症候群はオートファジーの欠陥に関連する多臓器障害であり、免疫系と脳や心臓などの臓器の解剖学的および機能的形成におけるオートファジー経路の基本的役割が示唆されている
臨床報告
本児は健康な近親のヤマニ両親の最初の製品である。 妊娠36週、体重3.2kg、頭囲31cm、アプガースコア8/8で、順調な妊娠期間を経て満床となった。 チアノーゼを伴い、生後から吸啜が悪く、授乳のたびに窒息発作を繰り返したが、残念ながら放置された経緯がある。 嘔吐や皮疹の既往はなく、異常な動きの既往もない。 家族歴に流産や死亡例はない。
診察では、顔色が悪く、悪液質で異形に見えた(図1)。 (皮膚,眼,金髪の色素沈着,広い額,多弁,長い口蓋垂,小顎症)。 体重は4,100g(3-10th centile)、体長は53cm(3-10th centile)、頭囲は33cm(3th centile)であった。 眼科検査では、軽度の水平眼振、両側白内障、フレア網膜を認めた。 心音は正常で雑音を認めず、深部低血圧であった
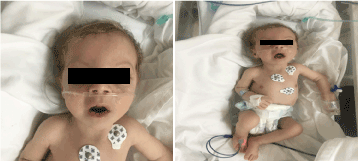
図1. 生後2ヶ月の臨床写真(a)低緊張、(b)金髪長顎、小顎症
検査では、小球性・低色素性貧血、腎臓プロファイル、凝固に異常はありませんでした。 アラニントランスアミナーゼ(ALT)は183-231(10-45 U/L),アスパラギン酸アミノトランスフェラーゼ(AST)は193-301(10-45 U/L)とそれぞれ高値であった。 ビリルビンは正常であった. CK:389U/L、LDH:612U/L。 アンモニアは13mmol/Lと正常であった. 免疫学的検査では、免疫グロブリン値の減少が認められたが、IgA: 0.1 g/L (0.7-4), IgG: 4.51 g/L (7-16), IgM: .8 g/L (0.4-2.3). 正常値内であった。 リンパ球サブセット(CD3、CD4、CD8、CD19、CD22)数は正常であった。 血液、尿、増殖はなかった。 血清学的検査では,トキソプラズマ,風疹,サイトメガロウイルス,単純ヘルペス,肝マーカーはすべて正常であった. 胸部レントゲンでは肺浸潤と心肥大を認めた. 腹部超音波検査で両側水腎症が認められた. 心エコーで左室肥大を指摘された. 脳の磁気共鳴画像(MRI)により脳梁の奇形が確認された(図2)。 遺伝子検査ではEPG5のホモ接合性機能喪失変異(EPG5:NM_020964:exon9:c.1924C>T:p.R642X)
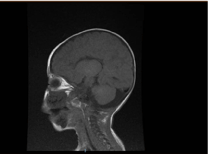
Figure 2. 脳のMRI、矢状面のT1強調断面で脳梁の奇形を示す
入院2週目に、人工呼吸を必要とする誤嚥性肺炎を繰り返し、急速に病状が悪化した。 8127>
材料と方法
WESはゲノムDNAに対してAgilent Sure Select Target Enrichmentワークフローを用いて行い、DNA断片ライブラリから関心領域を捕捉する。 イルミナHiSeq 2500シーケンスシステムで全エキソームのシーケンスを行い、ターゲット領域の95%を最低30Xのカバレッジでカバーする。 プロバンドのエクソームDNA配列はマッピングされ、ヒトゲノムビルドUCSC hg19参照配列と比較される。 サウジアラビア診断研究所では、社内パイプラインを使用してプロバンドの塩基配列を参照配列と比較する。 既知のタンパク質をコードするRefSeq遺伝子のターゲットとなるコードエクソンのカバレッジとクオリティの評価が実施されます。 エクソーム解析では、アラブ人集団用にカスタマイズされた独自のデータベースを使用して、プロバンドの数千もの遺伝的変異を調査します。 これらのサブセットは、American College of Medical Genetics and Genomics (ACMG) (Richards, et al. 2015) のガイドラインを使用して特性評価され、臨床的関連性が分類されます。 サンガーシークエンスは、特に明記しない限り、報告書に含まれるすべてのバリアントを検証するために行われる
結果
この患者では、EPG5のホモ接合性機能喪失バリアント(EPG5:NM_020964:exon9:c.1924C>T:p.R642X)が確認されました。 ACMGガイドライン(Gen in Med 17:5, 2015)によれば、それは病原性バリアントとして分類されるべきものである。 EPG5のホモ接合型Loss of Function(LOF)変異は、Vici症候群の一般的なメカニズムである242840 。
議論
Vici症候群は、常染色体劣性パターンで遺伝する珍しい先天性多系統疾患であり、成長不全、色素沈着、反復性感染症によって明らかな免疫異常、発達遅延、発作、白内障、心筋症および神経筋異常の神経異常を特徴とし、後者は最近の数例の報告にのみ記述されています 。 Vici症候群は1988年にDionisi Viciらによって、アルビニズム、ACC、白内障、心筋症、精神遅滞、免疫不全を呈する2人の兄弟の症例で初めて報告され、その後1999年にdel Campoらによって「Vici症候群」と命名されました。 Vici症候群は男女ともに観察され,EPG5遺伝子には多くの関連変異が同定されている。
我々の患者の両親は血縁関係にあり,Vici症候群の常染色体劣性遺伝を支持している。 31人の患者のうち、私たちを含めて13人は兄弟姉妹である。 Vici症候群は染色体18q12.3上のEPG5の劣性突然変異によるもので,EPG5は高等生物における重要なオートファジー制御因子であるectopic P granules protein 5 (EPG5)をコードしている。 オートファジーは、欠陥のあるタンパク質や小器官の除去、感染症に対する防御、変化する代謝需要への適応に重要な役割を持つ、進化を通じて保存されている基本的な細胞分解経路である。
Vici 症候群の患者における CNS 開発、免疫制御、皮膚色素形成は、より幅広い細胞プロセスにおけるオートファジーの関与が示唆されている。 脳では、オートファジーは様々な神経変性疾患における重要な発症メカニズムとして広く研究されている。
Vici症候群の兆候と症状のほとんどは出生時に存在する。白内障、心筋症、免疫不全などのいくつかの特徴は出生時に常に存在するとは限らず、最初の数年間で進化すると予想される . また、ほとんどの患者が心筋症や肺炎のために生後3年以内に死亡している
謝辞
著者は患者の両親とサウジアラビアの診断研究所に感謝します。
- Dionisi Vici C, Sabetta G, Gambarara M, Vigevano F, Bertini E, et al. (1988) 2人の兄弟における脳梁奇形、複合免疫不全、両白内障、色素欠乏症. Am J Med Genet 29:1-8.
- Thomas Cullup, Ay Lin Kho, Carlo Dionisi-Vici (2013) Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nature Genetics 45: 83-87.
- Said E, Soler D, Sewry C (2012) Vici syndrome-a rapidly progressive neurodegenerative disorder with hypopigmentation, immunodeficiency and myopathic changes on muscle biopsy.「Vici症候群-急速に進行する神経変性疾患-」. Am J Med Genet A 158: 440-444.
- del Campo M, Hall BD, Aeby A, Nassogne MC, Verloes A, et al. (1999) Albinism and agenesis of the corpus callosum with profound developmental delay.アルビニズムと脳梁閉鎖症。 Vici症候群、常染色体劣性遺伝の証拠。 Am J Med Genet 85: 479-485.
- Ehmke N, Parvaneh N, Krawitz P, Ashrafi MR, Karimi P, et al. (2014) First description of a patient with Vici syndrome due to a mutation affecting the penultimate exon of EPG5 and review of the literature.Vici症候群の患者を初めて報告した。 Am J Med Genet A 164A: 3170-3175.
- Al-Owain M, Al-Hashem A, Al-Muhaizea M, Humaidan H, Al-Hindi H, et al. (2010) Vici syndrome associated with unilateral lung hypoplasia and myopathy. Am J Med Genet A 152A: 1849-1853.
- Byrne S, Dionisi-Vici C, Smith L, Gautel M, Jungbluth H, et al. (2016) Vici syndrome: a review.「Vici症候群:レビュー」. Orphanet J Rare Dis 11: 21.
- Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, et al. (2006) Aggregate-prone proteins are cleared from the cytosol by autophagy: Therapeutic implications. Curr Top Dev Biol 76: 89-101.
- Cullup T, Dionisi-Vici C, Kho AL, Yau S, Mohammed S, et al. (2014) Clinical Utility gene card for.を参照。 Vici Syndrome. Eur J Hum Genet 22.
- Curtis Rogers R, Bridgette Aufmuth, Stephanie Monesson (2011) Vici Syndrome: 脳異常、心筋症、重度の知的障害を伴うまれな常染色体劣性症候群. 遺伝学のケースレポート2011。 421582.