Regardez les articles récents
Abstract
Objet : Rapporter un cas d’hypopigmentation oculocutanée trouvé chez un nourrisson avec une hypotonie sévère, une cardiomyopathie et une agénésie du corps.
Rapport de cas : Cette étude a concerné un bébé de sexe masculin qui a été accouché après une période de gestation de 37 semaines. Produit de parents Yamani consanguins en bonne santé avec un retard de croissance, des cataractes bilatérales congénitales et une dysmorphie faciale, des cheveux clairs et de la peau, un nystagmus et il avait une hypertrophie du ventricule gauche. Des mutations dans le gène epg5 ont été identifiées comme la cause du syndrome de Vici.
Conclusion : Le syndrome de Vici est une entité clinique distincte. Ses principales manifestations cliniques comprennent l’hypotonai, le retard de développement, l’albinisme, la cataracte, la cardiomyopathie, le déficit immunitaire et l’agénésie du corps calleux.
Mots clés
Syndrome de Vici ; agénésie du corps calleux ; EPG5
Introduction
Le syndrome de Vici est une maladie congénitale grave, à transmission récessive, caractérisée par les principales caractéristiques suivantes : agénésie du corps calleux (ACC), cataracte, hypopigmentation oculo-cutanée, cardiomyopathie, retard de croissance et variable.
Il a été décrit pour la première fois en 1988, chez deux frères présentant un syndrome malformatif composé d’une agénésie du corps calleux, d’une hypopigmentation cutanée, d’une cataracte bilatérale, d’une fente labiale et palatine et d’une immunodéficience combinée. Les frères et sœurs souffraient d’un retard psychomoteur grave, de crises d’épilepsie, d’infections respiratoires graves récurrentes et de candidose cutanéo-muqueuse chronique. Ils sont décédés de bronchopneumonie à l’âge de 2 et 3 ans .
Le syndrome de Vici est un trouble multisystémique associé à une autophagie défectueuse et suggère un rôle fondamental de la voie de l’autophagie dans le système immunitaire et la formation anatomique et fonctionnelle d’organes tels que le cerveau et le cœur .
Rapport clinique
Ce bébé est le premier produit de parents Yamani consanguins en bonne santé. Il était à terme, après une grossesse sans incident avec un accouchement normal du vertex à 36 semaines d’âge gestationnel avec un poids de naissance de 3,2 kg, une circonférence crânienne de 31 cm et un score d’Apgar de 8/8.
À 25 jours, il a été hospitalisé avec des antécédents de toux, d’essoufflement et de fièvre depuis trois jours. Associé à une cyanose, le patient avait des antécédents de mauvaise succion et des crises d’étouffement récurrentes à chaque tétée depuis la naissance, mais malheureusement, il a été négligé. Aucun antécédent de vomissement ou d’éruption cutanée et aucun antécédent de mouvement anormal. Pas d’antécédents familiaux d’avortements ou de décès.
À l’examen, il était pâle, cachectique et dysmorphique (figure 1). (peau hypopegmentée, œil et cheveux blonds, front large, hyperbolisme,long philtrum et micrognathie). Son poids était de 4 100 g (3-10e centile), sa longueur de 53 cm (3-10e centile) et son périmètre crânien de 33 cm (3e centile). Les examens ophtalmologiques ont révélé un léger nystagmus horizontal, une cataracte bilatérale et une rétine flasque. Plein de crépitation sur la poitrine bilatérale, pas de murmure avec le son du cœur normal et avait une hypotonie profonde.
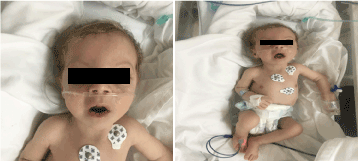
Figure 1. Photo clinique du patient à l’âge de 2 mois (a) Hypotonie, (b) blond de cheveux long philtrum et micrognathie
L’examen de laboratoire a révélé une anémie microcytaire et hypochrome, les profils rénaux et la coagulation étaient sans particularité. L’alanine transaminase (ALT) et l’aspartate aminotransférase (AST) étaient élevées à 183-231 (10-45 U/L), et 193-301 (10-45 U/L), respectivement. La bilirubine était normale. CK : 389 U/L et LDH : 612 U/L. L’ammoniac était normal 13 mmol/L. Le bilan immunologique a révélé des taux réduits d’immunoglobulines IgA : 0,1 g/L (0,7-4), mais les IgG : 4,51 g/L (7-16), IgM : .8 g/L (0,4-2,3). étaient dans les limites de la valeur normale. Le nombre de sous-ensembles de lymphocytes (CD3, CD4, CD8, CD19 et CD22) était normal. Le sang, l’urine et n’étaient pas en croissance. Le test sérologique pour la toxoplasme, la rubéole, le cytomégalovirus, l’herpès simplex, et les marqueurs hépatiques étaient tous normaux. La radiographie du thorax a montré une infiltration pneumonique et une cardiomégalie. L’échographie abdominale a révélé une hydronéphrose bilatérale. L’étude échocardiographique a permis de détecter une hypertrophie ventriculaire gauche. L’imagerie par résonance magnétique (IRM) du cerveau a confirmé une agénésie du corps calleux (figure 2). Les tests génétiques ont confirmé une variante homozygote de perte de fonction de l’EPG5 (EPG5:NM_020964:exon9:c.1924C>T:p.R642X)
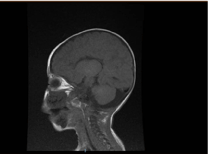
Figure 2. IRM du cerveau, coupe sagittale pondérée en T1 montrant une agénésie du corps calleux
Au cours de la 2e semaine d’admission, son état s’est rapidement détérioré avec des épisodes répétés de pneumonie par aspiration qui ont nécessité une ventilation. Finalement, le patient a fait un arrêt cardio-pulmonaire et est décédé.
Matériels et méthodes
La WES est réalisée sur de l’ADN génomique en utilisant le workflow d’enrichissement de cible Agilent Sure Select pour capturer les régions d’intérêt à partir d’une bibliothèque de fragments d’ADN. L’exome entier est séquencé sur le système de séquençage Illumina HiSeq 2500 avec une couverture minimale de 30X de 95% des régions cibles. Les séquences d’ADN de l’exome du proband sont cartographiées et comparées à la séquence de référence UCSC hg19 construite à partir du génome humain. Saudi Diagnostic Laboratories utilise un pipeline interne pour comparer la séquence du patient à la séquence de référence. Une évaluation de la couverture et de la qualité des exons codants ciblés des gènes RefSeq codant pour les protéines connues est effectuée. Les analyses de l’exome interrogent des milliers de variants génétiques chez le patient en utilisant des bases de données propriétaires adaptées aux populations arabes. Des sous-ensembles de ces variantes sont caractérisés en utilisant les directives de l’American College of Medical Genetics and Genomics (ACMG) (Richards, et al. 2015) pour classer leur pertinence clinique. Un séquençage Sanger est effectué pour valider tous les variants inclus dans le rapport, sauf indication contraire
Résultats
Un variant homozygote de perte de fonction d’EPG5 (EPG5:NM_020964:exon9:c.1924C>T:p.R642X) a été identifié chez ce patient. Selon les directives de l’ACMG (Gen in Med 17:5, 2015), il doit être classé comme un variant pathogène. Les variants homozygotes de perte de fonction (LOF) d’EPG5 sont un mécanisme commun du syndrome de Vici, 242840 .
Discussion
Le syndrome de Vici, est un trouble congénital rare de plusieurs systèmes qui est hérité dans un modèle autosomique récessif, il est caractérisé par un retard de croissance, une hypopigmentation, des défauts immunologiques évidents par des infections récurrentes, des anomalies neurologiques de retard de développement, des crises, des cataractes, une cardiomyopathie et des anomalies neuromusculaires ; avec le dernier étant seulement décrit dans quelques rapports de cas récents . Le syndrome de Vici a été rapporté pour la première fois en 1988 par Dionisi Vici, et al. dans le cas de deux frères présentant un albinisme, une ACC, des cataractes, une cardiomyopathie, un retard mental et une immunodéficience, et a ensuite été nommé « syndrome de Vici » par del Campo, et al. en 1999. Le syndrome de Vici a été observé chez les hommes et les femmes, et un certain nombre de mutations apparentées ont été identifiées dans le gène EPG5 .
Nos parents patients sont consanguins et soutiennent la transmission autosomique récessive du syndrome de Vici. Treize des 31 patients, dont le nôtre, sont des frères et sœurs. Des cas ont été rapportés dans lesquels les parents étaient des cousins éloignés .
Le syndrome de Vici est dû à des mutations récessives dans EPG5 sur le chromosome 18q12.3, codant pour la protéine 5 des granules P ectopiques (EPG5), un régulateur clé de l’autophagie dans les organismes supérieurs. L’autophagie est une voie de dégradation cellulaire fondamentale conservée tout au long de l’évolution avec des rôles importants dans l’élimination des protéines et des organites défectueux, la défense contre les infections et l’adaptation aux demandes métaboliques changeantes.
Le développement du SNC, la régulation immunitaire et la pigmentation de la peau chez les patients atteints du syndrome de Vici impliquent l’autophagie dans un plus large éventail de processus cellulaires. Dans le cerveau, l’autophagie a été largement étudiée comme un mécanisme pathogène clé dans divers troubles neurodégénératifs .
La plupart des signes et des symptômes du syndrome de vici sont présents à la naissance ; certaines caractéristiques comme la cataracte, la cardiomyopathie et l’immunodéficience ne sont pas toujours présentes à la naissance, mais devraient évoluer au cours des premières années . Et la plupart des patients sont décédés au cours des trois premières années de vie secondaire à une cardiomyopathie ou une pneumonie
Remerciements
Les auteurs souhaitent remercier les parents du patient et le laboratoire de diagnostic saoudien.
- Dionisi Vici C, Sabetta G, Gambarara M, Vigevano F, Bertini E, et al. (1988) Agénésie du corps calleux, immunodéficience combinée, cataracte bilatérale et hypopigmentation chez deux frères. Am J Med Genet 29:1-8.
- Thomas Cullup, Ay Lin Kho, Carlo Dionisi-Vici (2013) Des mutations récessives dans EPG5 causent le syndrome de Vici, un trouble multisystème avec autophagie défectueuse. Nature Genetics 45 : 83-87.
- Said E, Soler D, Sewry C (2012) Le syndrome de Vici-un trouble neurodégénératif rapidement progressif avec hypopigmentation, immunodéficience et changements myopathiques sur la biopsie musculaire. Am J Med Genet A 158 : 440-444.
- del Campo M, Hall BD, Aeby A, Nassogne MC, Verloes A, et al. (1999) Albinisme et agénésie du corps calleux avec retard profond du développement : Le syndrome de Vici, preuve d’une hérédité autosomique récessive. Am J Med Genet 85 : 479-485.
- Ehmke N, Parvaneh N, Krawitz P, Ashrafi MR, Karimi P, et al. (2014) Première description d’un patient atteint du syndrome de Vici dû à une mutation affectant l’avant-dernier exon d’EPG5 et revue de la littérature. Am J Med Genet A 164A : 3170-3175.
- Al-Owain M, Al-Hashem A, Al-Muhaizea M, Humaidan H, Al-Hindi H, et al. (2010) Syndrome de Vici associé à une hypoplasie pulmonaire unilatérale et à une myopathie. Am J Med Genet A 152A : 1849-1853.
- Byrne S, Dionisi-Vici C, Smith L, Gautel M, Jungbluth H, et al. (2016) Syndrome de Vici : une revue. Orphanet J Rare Dis 11 : 21.
- Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, et al. (2006) Aggregate-prone proteins are cleared from the cytosol by autophagy : therapeutic implications. Curr Top Dev Biol 76 : 89-101.
- Cullup T, Dionisi-Vici C, Kho AL, Yau S, Mohammed S, et al. (2014) Carte de gènes d’utilité clinique pour : Le syndrome de Vici. Eur J Hum Genet 22.
- Curtis Rogers R, Bridgette Aufmuth, Stephanie Monesson (2011) Syndrome de Vici : Un syndrome autosomique récessif rare avec des anomalies cérébrales, une cardiomyopathie et une déficience intellectuelle sévère. Case Reports in Genetics 2011 : 421582.