Közvetlen Xa-gátlók felfedezése és fejlesztése
Xa faktor: Szerkezet és kötőhelyekSzerkesztés

A IIa, Xa, VIIa, IXa és XIa faktorok mind proteolitikus enzimek, amelyeknek specifikus szerepük van a véralvadási kaszkádban. A Xa faktor (FXa) a legígéretesebb, mivel az intrinsic és extrinsic útvonal metszéspontjában helyezkedik el, valamint minden egyes Xa molekulára körülbelül 1000 trombinmolekulát termel, ami erős antikoaguláns hatást eredményez. Az FXa az FX-ből egy 52 aminosavból álló aktiváló peptid hasításával keletkezik, mivel az “a” az Xa faktorban aktiváltat jelent. Az FXa 254 aminosavból álló katalitikus doménből áll, és egy 142 aminosavból álló könnyűlánchoz is kapcsolódik. A lánc tartalmaz GLA-domént és két epidermális növekedési faktor domént (EGF-szerű domént).
Az FXa aktív helye úgy van felépítve, hogy fiziológiás szubsztrátok hasítását katalizálja, és a prothrombinban PhePheAsnProArg-ThrPhe és TyrIleAspGlyArg-IleVal domént hasít. Az FXa négy úgynevezett zsebbel rendelkezik, amelyek a Xa faktorhoz kötődő szubsztrátok célpontjai. Ezeket a zsebeket különböző aminosavak szegélyezik, és az Xa-inhibitorok ezeket a zsebeket célozzák meg, amikor az Xa faktorhoz kötődnek. Az Xa-inhibitorok affinitása és szelektivitása szempontjából a két legfontosabb zseb az S1 és az S4.
S1: Az S1 zseb egy hidrofób zseb, és egy aszparaginsav-maradékot (Asp-189) tartalmaz, amely egy bázikus csoport felismerőhelyeként szolgálhat. Az FXa az S1 zsebben egy maradék térrel rendelkezik, amelyet a Tyr-228, Asp-189 és Ser-195 maradékok bélelnek.
S2: Az S2 zseb egy kicsi és sekély zseb. Összeolvad az S4 zsebbel, és kis aminosavaknak van helye. Úgy tűnik, hogy a Tyr-99 blokkolja a hozzáférést ehhez a zsebhez, így ez a zseb nem olyan fontos, mint az S1 és S4.
S3: Az S3 zseb az S1 zseb peremén helyezkedik el, lapos és az oldószernek kitett. Ez a zseb nem olyan fontos, mint az S1 és S4.
S4: Az S4 zseb hidrofób jellegű, és a zseb padlóját Trp-215 maradék alkotja. Az FXa Phe-174 és Tyr-99 maradékai a Trp-215-höz csatlakozva egy aromás dobozt alkotnak, amely képes alifás, aromás és pozitív töltésű fragmentumok megkötésére. A pozitív töltésű egységekhez való kötődés miatt kationlyukként írható le.
A direkt Xa-gátlók kémiai szerkezete és tulajdonságaiSzerkesztés
| Rivaroxaban | Apixaban | Edoxaban | ||
|---|---|---|---|---|
| MW (g/mol) | 436 | 460 | 548 | |
| Molekulaáris képlet | C19H18ClN3O5S | C25H25N5O4 | C24H30ClN7O4S | |
| alak | L | L | L | L |
| Ki | 0.4 nM | 0,08 nM | 0,561 nM | |
| IC50 | 0.7 nM | N/A | N/A | |
| Orális biológiai hozzáférhetőség (%) | 66-100 (dózis-függő) | 50 | 62 |

Az Xa-gátlók kötődése a Xa faktorhozSzerkesztés
A Xa-gátlók mindegyike úgynevezett L-alakban kötődik a Xa faktor aktív helyén belül. A Xa faktor kulcsfontosságú alkotóelemei az S1 és S4 kötőhelyek. Először azt figyelték meg, hogy a természetes vegyületek, az antistasin és a TAP, amelyek erősen poláros és ezért töltött komponensekkel rendelkeznek, bizonyos specificitással kötődnek a célponthoz. Ezért az újabb gyógyszereket pozitív töltésű csoportokkal tervezték, de ezek rossz biohasznosulást eredményeztek. A manapság forgalmazott Xa-gátlók ezért egy aromás gyűrűt tartalmaznak, amelyhez különböző részek kapcsolódnak az S1 és S4 kötőhelyekkel való különböző kölcsönhatások érdekében. Ez biztosítja a jó biológiai hozzáférhetőséget és a szilárd kötőerő fenntartását is. A jelenleg forgalomban lévő Xa-inhibitorok ezért az erősen poláris kölcsönhatások helyett a hidrofób és hidrogénkötésekre támaszkodnak.
Antistasin kötődése a Xa faktorhozSzerkesztés
Az Antistasin egy N- és egy C-terminális domént tartalmaz, amelyek aminosav-szekvenciája hasonló, ~40%-os azonossággal és ~56%-os homológiával. Mindkettő rövid β-lemez szerkezetet és 5 diszulfidkötést tartalmaz. Csak az N-terminális domén szükséges a Xa gátlásához, míg a C-terminális domén nem járul hozzá a gátló tulajdonságokhoz a 3 dimenziós szerkezet különbségei miatt, annak ellenére, hogy a C-terminális domén erősen analóg mintázatot mutat a tényleges aktív centrummal.
Az antistasin és az FXa kölcsönhatásában az FXa aktív centruma és inaktív felülete is részt vesz. Az antisztaszin N-terminális doménben lévő Arg-34 és Val-35 által alkotott reaktív helye megfelel az FXa kötőhelyének, valószínűleg az S1 zsebnek. Ugyanakkor az antisztázin reaktív helyén kívül elhelyezkedő Glu-15 az FXa felszínének pozitív töltésű maradékaihoz illeszkedik. A többszörös kötődés termodinamikailag előnyös és szubnanomoláris gátláshoz vezet (Ki = 0,3-0,6 nM).
A DX-9065a kötődése a Xa faktorhozSzerkesztés
A DX-9065a, az első kis molekulájú közvetlen Xa-gátló, egy amidinoaril-derivátum, amelynek molekulatömege 571,07g/mol. Pozitív töltésű amidinonaftalin csoportja sóhidat képez az FXa S1 zsebében lévő Asp-189 maradékhoz. A pirrolidingyűrű az FXa S4 zsebében a Tyr-99, Phe-174 és Trp-215 között illeszkedik.
A régebbi gyógyszerekkel, pl. a heparinnal ellentétben a DX-9065a szelektív az FXa számára a trombinhoz képest, annak ellenére, hogy az FXa és a trombin hasonló szerkezetűek. Ezt a homológ 192-es pozíciójában lévő aminosavmaradékban lévő különbség okozza. Míg az FXa egy glutamin-maradékot tartalmaz ebben a pozícióban, addig a trombin egy glutaminsavat, amely elektrosztatikus taszítást okoz a DX-9065a karboxilcsoportjával. Ezenkívül a trombin Glu-97-je és a DX-9065a pirrolidingyűrűjében rögzített amidincsoport közötti sóhíd csökkenti a DX-9065a molekula rugalmasságát, amely most nem tud eléggé elfordulni ahhoz, hogy elkerülje az elektrosztatikus ütközést. Ezért a trombin IC50 értéke >1000µM, míg az FXa IC50 értéke 0,16µM.

A rivaroxaban kötődése a Xa faktorhozSzerkesztés
A rivaroxaban FXa-hoz való kötődése a Gly-219 aminosavhoz kötött két hidrogénkötésen keresztül közvetít. Ez a két hidrogénkötés fontos szerepet játszik abban, hogy a hatóanyagot az FXa S1 és S4 szubhelyeire irányítsa. Az első hidrogénkötés egy erős kölcsönhatás, amely a rivaroxaban oxazolidinon magjának karbonil oxigénjétől származik. A második hidrogénkötés egy gyengébb kölcsönhatás, és a klorotiofén-karboxamid-rész aminocsoportjától származik.
Ez a két hidrogénkötés eredményezi, hogy a hatóanyag L-alakot alkot, és illeszkedik az S1 és S4 zsebekbe. A Phe-174, Tyr-99 és Trp-215 aminosavmaradványok egy keskeny hidrofób csatornát képeznek, amely az S4 kötőzseb. A rivaroxaban morfolinon része a Tyr-99 és a Phe-174 aminosavak közé “szorul”, és a rivaroxaban arilgyűrűje a Trp-215-re merőlegesen helyezkedik el. A morfolinon-karbonilcsoportnak nincs közvetlen kölcsönhatása az FXa gerincoszlopával, ehelyett hozzájárul a morfolinongyűrű planarizációjához, és ezért támogatja, hogy a rivaroxaban a két aminosav közé szoruljon.
A tiofénrész klór-szubsztituensének és a Tyr-228 aromás gyűrűjének kölcsönhatása, amely az S1 alján helyezkedik el, nagyon fontos, mivel kiküszöböli az erősen bázikus csoportok szükségességét az FXa iránti magas affinitáshoz. Ez teszi lehetővé, hogy a rivaroxaban, amely nem bázikus, jó orális biológiai hozzáférhetőséget és hatékonyságot érjen el.

Az apixaban kötődése a Xa faktorhozSzerkesztés
Az apixaban hasonló kötődési módot mutat, mint a rivaroxaban, és szoros inhibitor-enzim komplexet alkot, amikor az FXa-hoz kapcsolódik. Az apixaban p-metoxicsoportja az FXa S1 zsebéhez kapcsolódik, de úgy tűnik, hogy nincs kölcsönhatása az FXa e régiójának egyetlen maradékával sem. Az apixaban pirazol N-2 nitrogénatomja kölcsönhatásba lép a Gln-192-vel, a karbonil oxigén pedig a Gly-216-tal. Az apixaban fenil-laktámcsoportja a Tyr-99 és a Phe-174 között helyezkedik el, és orientációja miatt képes kölcsönhatásba lépni az S4 zseb Trp-215-ével. A laktámrész karbonil oxigéncsoportja egy vízmolekulával lép kölcsönhatásba, és úgy tűnik, hogy nem lép kölcsönhatásba az S4 zseb egyetlen maradékával sem.

Szerkezet-aktivitás-reláció (SAR)Edit
A bizonyos célpont ideális inhibitorának számító vegyület tervezésének fontos része, hogy megértsük a célpont aminosavsorrendjét, amelyhez a vegyületnek kötődnie kell. Mind a protrombin, mind az FXa modellezése lehetővé teszi a különbség levezetését és az egyes kötőhelyek aminosavainak azonosítását. Az FXa S1 zsebének alján a kötő aminosav az Asp-189, amelyhez amidinrészek tudnak kötődni. Az FXa kötőhelyének röntgenvizsgálata után kiderült, hogy az S1 zseb sík alakú, ami azt jelenti, hogy egy lapos amidinoaril-csoportnak sterikus akadály nélkül kellene kötődnie hozzá.
A modern direkt Xa-gátlók L alakú molekulák, amelyek végei tökéletesen illeszkednek az S1 és S4 zsebekbe. Az L-alak hosszú oldalának meg kell felelnie egy nagyon specifikus alagútnak a célpontok aktív helyén belül. Ennek elérése érdekében a molekuláknak ezt a részét úgy tervezték, hogy abban a régióban kevés formális kölcsönhatás legyen az FXa-val. Mivel nincs specifikus kötés, ezeknek a szereknek az FXa zsebei közötti illeszkedése növeli a gyógyszerek teljes specifitását az FXa molekulához. Az FXa S1 zsebe és az inhibitor közötti kölcsönhatás lehet ionos vagy nem ionos is, ami azért fontos, mert lehetővé teszi a rész kialakítását az orális biológiai hozzáférhetőség növelése érdekében. A korábban tervezett vegyületek töltött molekulák voltak, amelyek nem szívódtak fel jól a gyomor-bélrendszerben, és ezért nem értek el magas szérumkoncentrációt. Az újabb gyógyszereknek jobb a biológiai hasznosulása, mivel nem töltöttek, és nem ionos kölcsönhatásuk van az S1 zsebhez.
Rivaroxaban
A rivaroxaban SAR-fejlesztése során a kutatók rájöttek, hogy az oxazolidonin maghoz egy 5-klórtiofén-2-karboxamid csoport hozzáadása 200-szorosára növelheti a potenciát, ami korábban túl gyenge volt az orvosi alkalmazáshoz. E felfedezés mellett megerősítették az (S)-konfiguráció egyértelmű preferenciáját. Ez a vegyület ígéretes farmakokinetikai profillal rendelkezett, és nem tartalmazott erősen bázikus amidincsoportot, amelyet azonban korábban fontosnak tartottak az S1 zsebbel való kölcsönhatás szempontjából. Ezek az eredmények kiterjedt SAR (szerkezet-aktivitás kapcsolat) kutatásokhoz vezettek. A SAR-vizsgálatok során az R1-et határozták meg a hatásosság szempontjából legfontosabb csoportként. A pirrolidinon volt az első R1 funkciós csoport, amely jelentősen növelte a hatékonyságot, de a további kutatások még nagyobb hatékonyságot mutattak ki a morpholinon csoport helyett. Az R2 és R3 csoportokhoz hidrogén vagy fluor kapcsolódott, és gyorsan megállapították, hogy a hidrogén a legnagyobb hatékonyságot eredményezte. Az R2 és R3 csoportokat ezután különböző csoportokkal helyettesítették, amelyek mind kevésbé voltak hatásosak, mint a hidrogén, így a hidrogén lett a végeredmény. Mivel a klórtiofén-rész vízben való oldhatósága nem volt megfelelő, megpróbálták más csoporttal helyettesíteni, de nem jártak sikerrel. A klórtiofén-rész az S1 zseb alján lévő Tyr-228-hoz kötődik, ami kulcsfontosságú tényezővé teszi az FXa-hoz való kötődést illetően. A rivaroxaban egyszerre rendelkezik magas affinitással és jó biológiai hozzáférhetőséggel.

Apixaban

Az apixaban SAR fejlesztése során három csoportot kellett tesztelni a maximális hatásfok és biológiai hozzáférhetőség elérése érdekében. Az első tesztelendő csoport a nem aktív hely volt, mivel azt stabilizálni kell a p-metoxifenilcsoporton (S1 kötő rész) végzett SAR-vizsgálat előtt. Számos olyan csoport van, amely növeli a vegyület hatékonyságát, főként amidok, aminok és tetrazolok, de metilszulfonil- és trifluorometilcsoportok is. E csoportok közül a karboxamidnak van a legnagyobb kötődése, és hasonló alvadási aktivitást mutatott, mint a vegyületek.
Kutyás vizsgálatokban ez a karboxamidcsoporttal rendelkező, 13F nevű vegyület nagyszerű farmakokinetikai profilt, alacsony clearance-t, megfelelő felezési időt és megoszlási térfogatot mutatott. A stabilizáló csoport megtalálásának sikere miatt az S1 kötésrész (p-metoxi-fenil) SAR-kutatását abbahagyták. Az S4 kötőcsoportban az N-metilacetil és laktám analógok nagyon magas kötési affinitást mutattak az FXa-hoz, nagyszerű alvadást és szelektivitást mutattak más proteázokkal szemben. Az orientáció fontosnak bizonyult, mivel az N-metil-acetil az acetamidhoz képest 300-szor kisebb kötődést mutatott az FXa-hoz az S4 régió kötőhelyéhez közeli kedvezőtlen planaritás miatt.
SynthesisEdit
RivaroxabanEdit
A rivaroxaban kémiailag az n-aryloxazolidinonok csoportjába tartozik. E csoportba tartozó más gyógyszerek a linezolid és a tedizolid, mindkettő antibiotikum. Az n-aryloxazolidinonok szintézisét O-szilil védett etil(2,3-dihidroxipropil)-karbamátból kiindulva 2016-ban publikálták. Az egytégelyes reakcióban a karbamát enyhén bázikus körülmények között 2-oxazolidon gyűrűvé ciklizálódik, miközben az oxazolidon nitrogénje rézkatalizálással egyidejűleg arilizálódik. Különösen a rivaroxaban esetében a 3-morfolinon rézkatalizálással helyettesíti a benzolgyűrű p-pozíciójában lévő jódot. Ezt követően a szilil védőcsoportot eltávolítják, és a keletkező alkoholt aminocsoportra cserélik, amelyet az utolsó lépésben acileznek.
A rivaroxaban ipari előállítását a Bayer Healthcare 2005-ben szabadalomként bejegyezte. Ez N-(4-aminofenol)-morfolinonból indul ki, amelyet egy propilén-oxid származékkal alkileznek, amely egy ftalimid védőcsoportban részt vevő primer amint is tartalmaz. Ezután egy foszgén-egyenértéket adnak hozzá a 2-oxazolidon gyűrű kialakításához, majd eltávolítják a ftalimidet. A szabad amin most már acilálható, ami a rivaroxabant eredményezi.
A szabadalom szerint azonban a szintézisnek “különböző hátrányai vannak a reakcióvezetésben, ami különösen kedvezőtlen hatással van a készítményre”. A szabadalom egy másik, klórtiofén-származékból kiinduló szintézist is ismertet, amely alkalmasabb lenne az ipari eljárásra, de rámutat arra, hogy a végtermékből el kell távolítani a mérgező oldószereket vagy reagenseket. Ezért ez az út nem jelent alternatívát.
A rivaroxaban különböző más szintézisútjait is leírták.
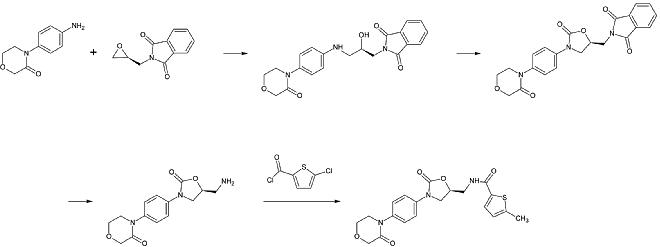
1. lépés: Elsődleges aromás amin alkilezése
2. lépés: A 2-oxazolinidon gyűrű képzése, foszgén-egyenérték felhasználásával
3. lépés: A ftalimid védőcsoport eltávolítása
4. lépés: A primer amin acilezése
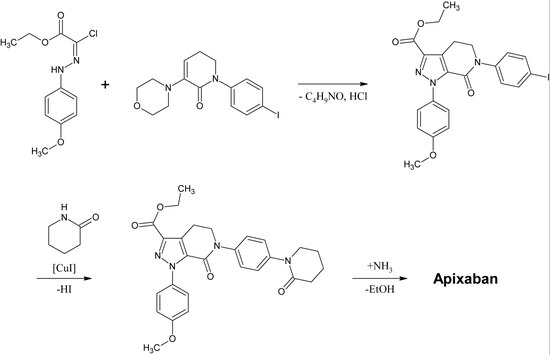
ApixabanSzerkesztés
Az apixaban első teljes szintézisét 2007-ben tették közzé. A reakció kulcslépése egy p-metoxifenil-klórhidrazon-származék és egy p-jódfenil-morfolin-dihidropiridin-származék (3+2)-cikloaddíciója. A HCl és a morfolin ezt követő eliminációja után a jódot rézkatalizálással 2-piperidinonnal helyettesítjük, az etil-észter pedig amiddá alakul (aminolízis). Ezt a reakciót 2009-ben szabadalomként bejegyezték.