Diagnostyka obrazowa nieprawidłowości istoty białej | Wrodzona hipomielinizacja mózgu; sieć dla choroby Pelizaeusa-Merzbachera i zaburzeń pokrewnych
Diagnostic Imaging of White Matter Abnormalities
Junichi TAKANASHI, Department of Pediatrics, Tokyo Women’s Medical University, Yachiyo Medical Center
Wprowadzenie
W tym artykule przedstawiam podejście stosowane w diagnostyce zaburzeń, które pojawiają się jako nieprawidłowe sygnały w istocie białej mózgu na obrazowaniu rezonansu magnetycznego (MRI), od obrazowania do diagnozy. Zaburzenia, które dotyczą głównie istoty białej, są ogólnie określane jako „leukoencefalopatia” lub „zaburzenia istoty białej” w języku angielskim.1, 2) 3) Inny termin, leukodystrofia, jest czasami mylony ze zwyrodnieniem istoty białej, ale w rzeczywistości odnosi się on do węższego spektrum zaburzeń z komponentem genetycznym (dziedziczne zaburzenia demielinizacyjne).
Oparta na obrazowaniu klasyfikacja zaburzeń istoty białej
Pojawienie się MRI radykalnie poprawiło naszą zdolność do wykrywania zmian w istocie białej ośrodkowego układu nerwowego. Wiele znanych form zaburzeń istoty białej wykazuje specyficzne objawy w MRI, co jest przydatne w ich diagnostyce. Identyfikacja wzorców nieprawidłowości w istocie białej widocznych w MRI (obrazowanie T1-zależne, T2-zależne lub FLAIR) ułatwia zawężenie możliwości w wielu rozpoznaniach różnicowych. Praktyczną wartość ma klasyfikacja Schiffmanna i van der Kampa dotycząca zaburzeń istoty białej w zależności od wyników MRI2, 4) (ryc. 1, tab. 1). Nawet jeśli nie prowadzi to do ostatecznego rozpoznania, klasyfikacja wyników obrazowania może prowadzić do późniejszego odkrycia nowego zaburzenia. Poniżej opisuję zaburzenia istoty białej w kategoriach powyższych klasyfikacji MRI i wyjaśniam główne typy zaburzeń.
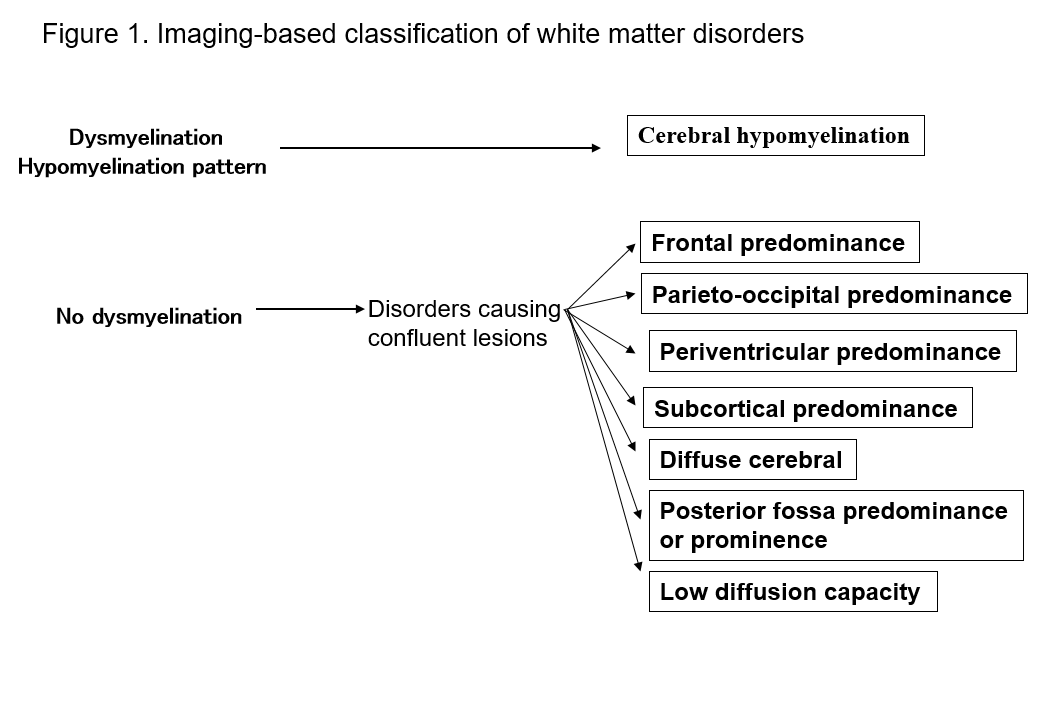
Tabela 1. Lista zaburzeń według wzorców MRI
- Przewaga czołowa
Choroba Alexandra, wariant czołowy adrenoleukodystrofii związanej z chromosomem X (ALD), leukodystrofia metachromatyczna (MLD), leukodystrofia neuroaksonalna ze sferoidami. - Przewaga parieto-occipitalna
X-linked adrenoleukodystrophy (ALD), Krabbe disease,early onset peroxisomal disorders, neonatal hypoglycemia. - Przewaga okołokomorowa
Leukodystrofia metachromatyczna (MLD), choroba Krabbego, zespół Sjögrena-Larssona, choroba ciałek poliglukozanowych u dorosłych, leukoencefalopatia z zajęciem pnia mózgu i rdzenia kręgowego oraz podwyższeniem stężenia mleczanów (LBSL), leukomalacja okołokomorowa (PVL), encefalopatia HIV, lipofuscynozy neuronalne o późniejszym początku. - Przewaga podkorowa
Kwasica L-2-hydroksyglutarowa, galaktozemia, zespół Kearns-Sayer, akadamia propionowa, zaburzenia cyklu mocznikowego, choroba Canavana. - Rozlana mózgowa
leukoencefalopatia megalencefaliczna z torbielami podkorowymi (MLC), leukoencefalopatia ze zanikającą istotą białą (VWM), wrodzona dystrofia mięśniowa z niedoborem merozyny, choroba mitochondrialna, niedobór kofaktora molibdenu, niedobór oksydazy siarczynowej, zaawansowane przypadki zaburzeń istoty białej. - Przewaga lub uwypuklenie tylnego dołu czaszki
Przepukliny móżdżku i szypułek móżdżku: ksantomatoza cerebrotendynowa (CTX), zaburzenia peroksysomalne, choroba Alexandra, leukoencefalopatia z zajęciem pnia mózgu i rdzenia kręgowego oraz podwyższeniem stężenia mleczanów (LBSL), choroba moczu wywołana syropem klonowym, histiocytoza, autosomalna dominująca leukodystrofia dorosłych związana z duplikacją lamin B1, toksyczność heroiny i kokainy.
Zmiany w pniu mózgu: Choroba Alexandra, LSBL, zaburzenia peroksysomalne, choroba Wilsona, choroba dorosłych poliglukozanów, zespół Leigha, zanik dentatorubropallidoluysian (DRPLA), choroba dorosłych ciał poliglukozanowych, dorosła autosomalna dominująca leukodystrofia związana z duplikacją lamin B1. - Zmiany wieloogniskowe
Zespół TORCH (wrodzone zakażenie cytomegalowirusem), bruceloza, ostre rozsiane zapalenie mózgu i rdzenia kręgowego (ADEM), stwardnienie rozsiane (SM), neuromyelitis optica (NMO), mózgowa autosomalnie dominująca arteriopatia z zawałami podkorowymi i leukoencefalopatią (CADASIL), miażdżyca, angiopatia amyloidowa, choroba małych naczyń mózgowych związana z COL4A1, choroba Fabry’ego, zespół Susaca, choroba mitochondrialna, kwasica L-2-hydroksyglutarowa, mukopolisacharydoza (MPS), nieprawidłowości chromosomalne (takie jak zespół 6p). - Zmiany o małej zdolności dyfuzyjnej
Choroba moczowa wywołana syropem klonowym, niedobór adenozylotransferazy metioninowej I/III, fenyloketonuria, hiperglikemia nieketotyczna, choroba Canavana, aktywne zmiany w chorobie Krabbego i leukodystrofia metachromatyczna.
1. Hipomielinizacja istoty białej mózgu
Odnosi się do grupy zaburzeń, w których tworzenie osłonki mielinowej jest upośledzone lub opóźnione, a ich obrazy przypominają obrazy noworodków z niedojrzałą mielinizacją. Na obrazach T2-ważonych istota biała charakterystycznie pojawia się jako rozległa hiperintensywność, która jest słabo zaznaczona w porównaniu z korą. Więcej szczegółów można znaleźć na stronie wrodzonej hipomielinizacji mózgu.
Jeśli zmiany w istocie białej nie są zgodne z hipomielinizacją istoty białej mózgu, należy ustalić, czy są one konfluentne czy mnogie.2) Konfluentne zmiany w istocie białej są zwykle spowodowane dziedzicznym zwyrodnieniem istoty białej (leukodystrofią) i w większości przypadków są obustronnie symetryczne. Mnogie zmiany w istocie białej są zwykle asymetryczne i nabyte. Konfluentne zmiany w istocie białej są dalej podzielone na kategorie 2-7 poniżej.
2. Przewaga czołowa
W tej grupie zaburzeń, rozległe zmiany w istocie białej są obecne głównie w płatach czołowych. Obejmują one chorobę Alexandra, wariant czołowy adrenoleukodystrofii X (ALD), leukodystrofię metachromatyczną (MLD) i leukodystrofię neuroaksonalną ze sferoidami.
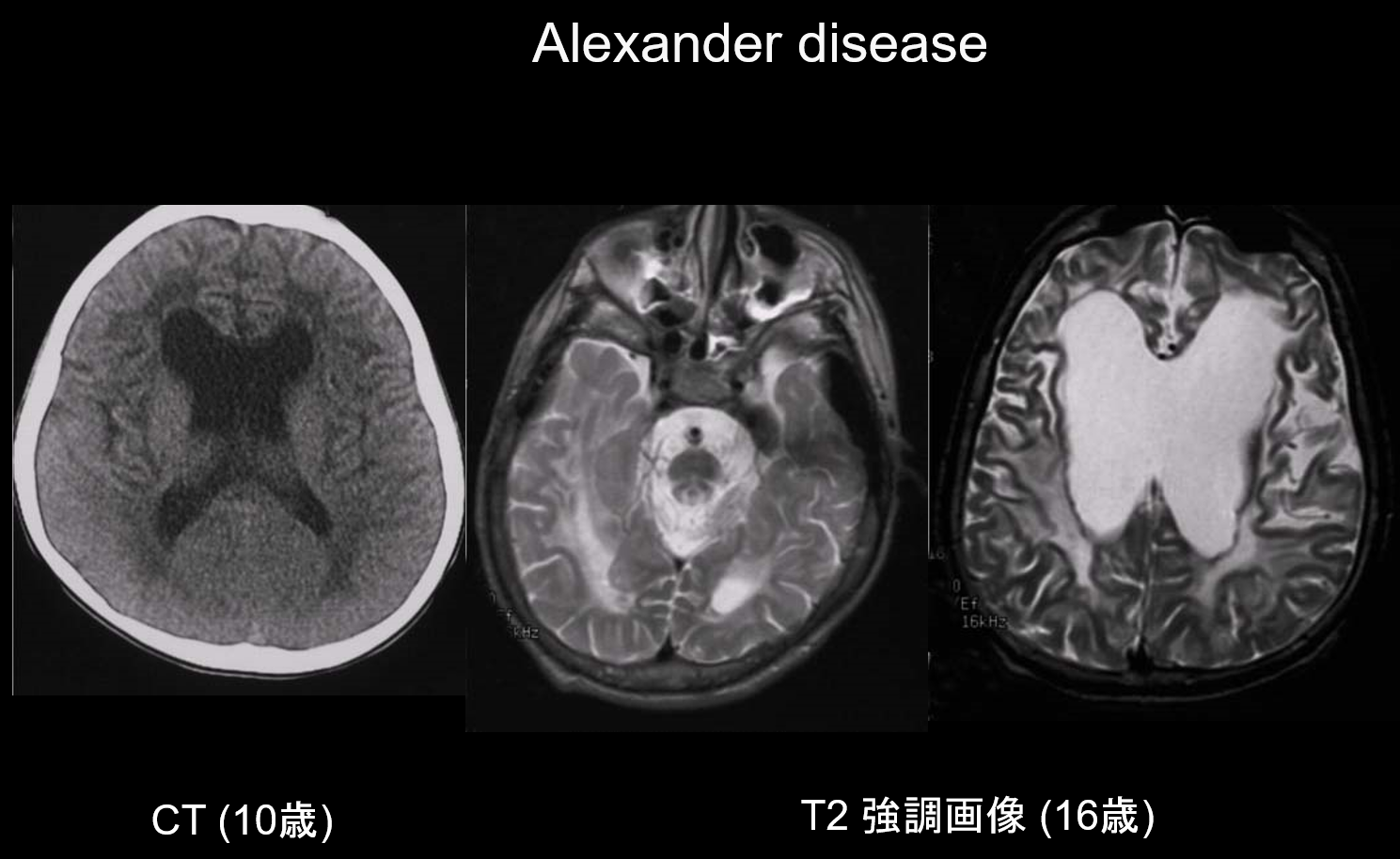
(a) Choroba Alexandra.
Choroba Alexandra jest dziedziczonym autosomalnie dominująco zaburzeniem spowodowanym mutacją w genie GFAP na chromosomie 17q21. Powoduje ona nagromadzenie włókien Rosenthala w komórkach glejowych gwiaździstych. Włókna te zbudowane są z GFAP i białek stresu (αB-krystalina i HSP27). Choroba Alexandra występuje głównie w wieku niemowlęcym, między 3 miesiącem a 2 rokiem życia, objawiając się megalencefalią, opóźnieniem rozwoju, porażeniem spastycznym i padaczką. W MRI może wykazywać: (i) rozległe zmiany w istocie białej, głównie w płatach czołowych; (ii) hiperintensywne T1 i hipointensywne T2 wokół komór bocznych; (iii) zmiany w zwojach podstawy i wzgórzu; (iv) zmiany w pniu mózgu; oraz (v) wzmocnienie kontrastowe aktywnych zmian (Rycina 2). We wczesnych stadiach, wraz ze zmianami w istocie białej i putamen, widoczny jest obrzęk, który może stopniowo powodować zanik lub tworzenie się torbieli.
3. Przewaga okołopotyliczna
Główną cechą tej grupy zaburzeń są zmiany w istocie białej okołopotylicznej. Obejmują one adrenoleukodystrofię (ALD) sprzężoną z chromosomem X, chorobę Krabbego, zaburzenia peroksysomalne o wczesnym początku oraz hipoglikemię noworodkową.
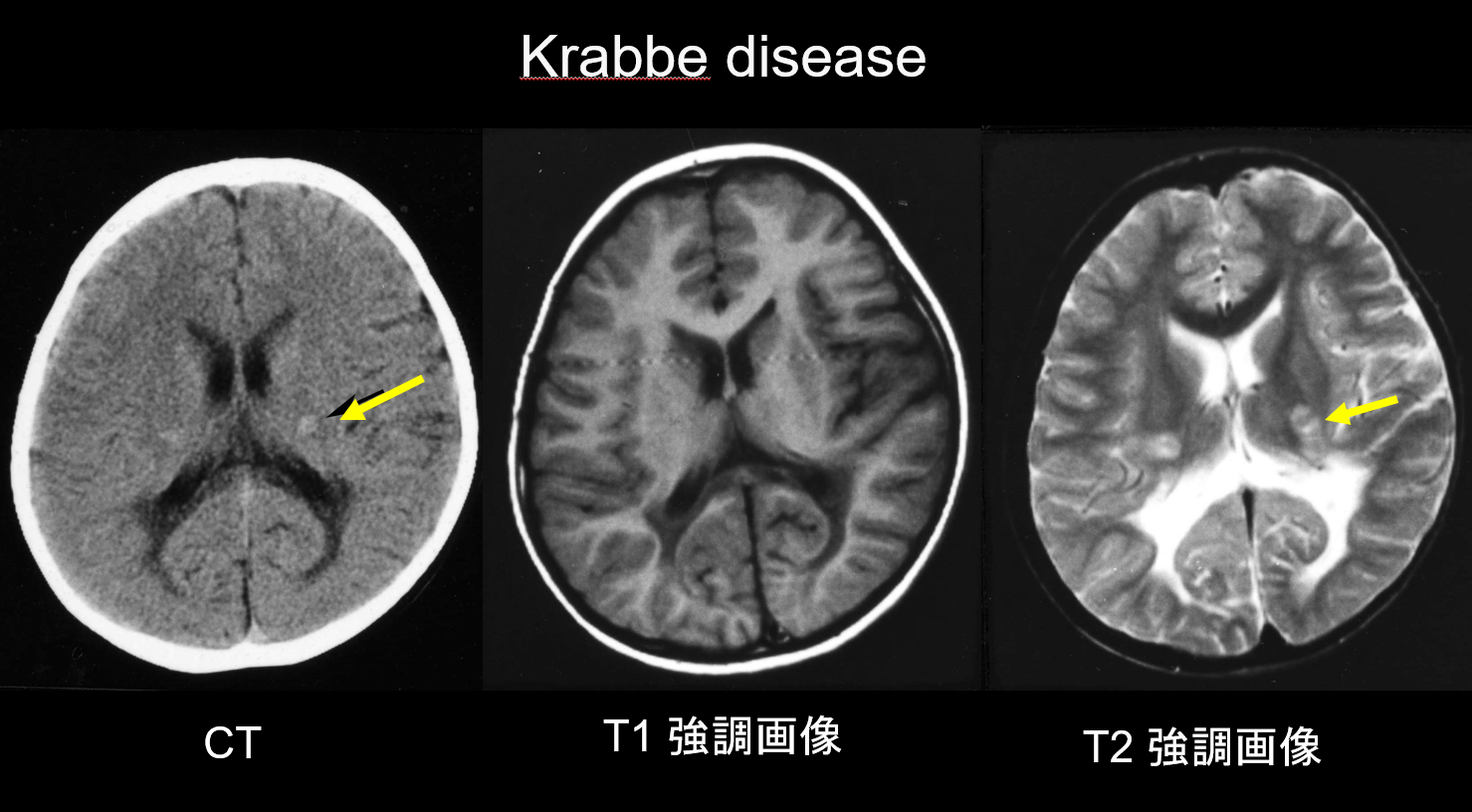
(a) Choroba Krabbego.
Choroba Krabbego jest dziedziczonym autosomalnie recesywnie zaburzeniem (lizosomalna choroba spichrzeniowa) spowodowanym niedoborem galaktozyloceramidazy (chromosom 14q31), w którym akumulacja wysoce cytotoksycznej psychozyny jest uważana za przyczynę rozległej demielinizacji. Pojawiają się również duże, wielojądrowe komórki zwane „komórkami globoidalnymi”. W zależności od wieku, w którym się pojawia, jest klasyfikowana jako choroba niemowlęca, późnoniemowlęca, młodzieńcza lub dorosłych. Większość przypadków ma charakter niemowlęcy i zaczyna się od wystąpienia gorączki, drażliwości, trudności w karmieniu, opóźnienia rozwoju, neuropatii obwodowej, spastyczności i zaniku nerwu wzrokowego w wieku 3-6 miesięcy. We wczesnych stadiach tomografia komputerowa (CT) ujawnia charakterystyczną hiperdensyjność we wzgórzu i koronie promienistej. Uważa się, że odzwierciedla to komórki globoidalne o dużej gęstości i proliferację gleju. Rezonans magnetyczny może również wykazać hiperintensywność T1 i hipointensywność T2 wokół komór, jak również struktury liniowe podobne do tych obserwowanych w MLD (ryc. 3). Jądro zębate móżdżku, istota biała móżdżku i drogi piramidowe pnia mózgu wykazują hiperintensywność T2 od wczesnego stadium.
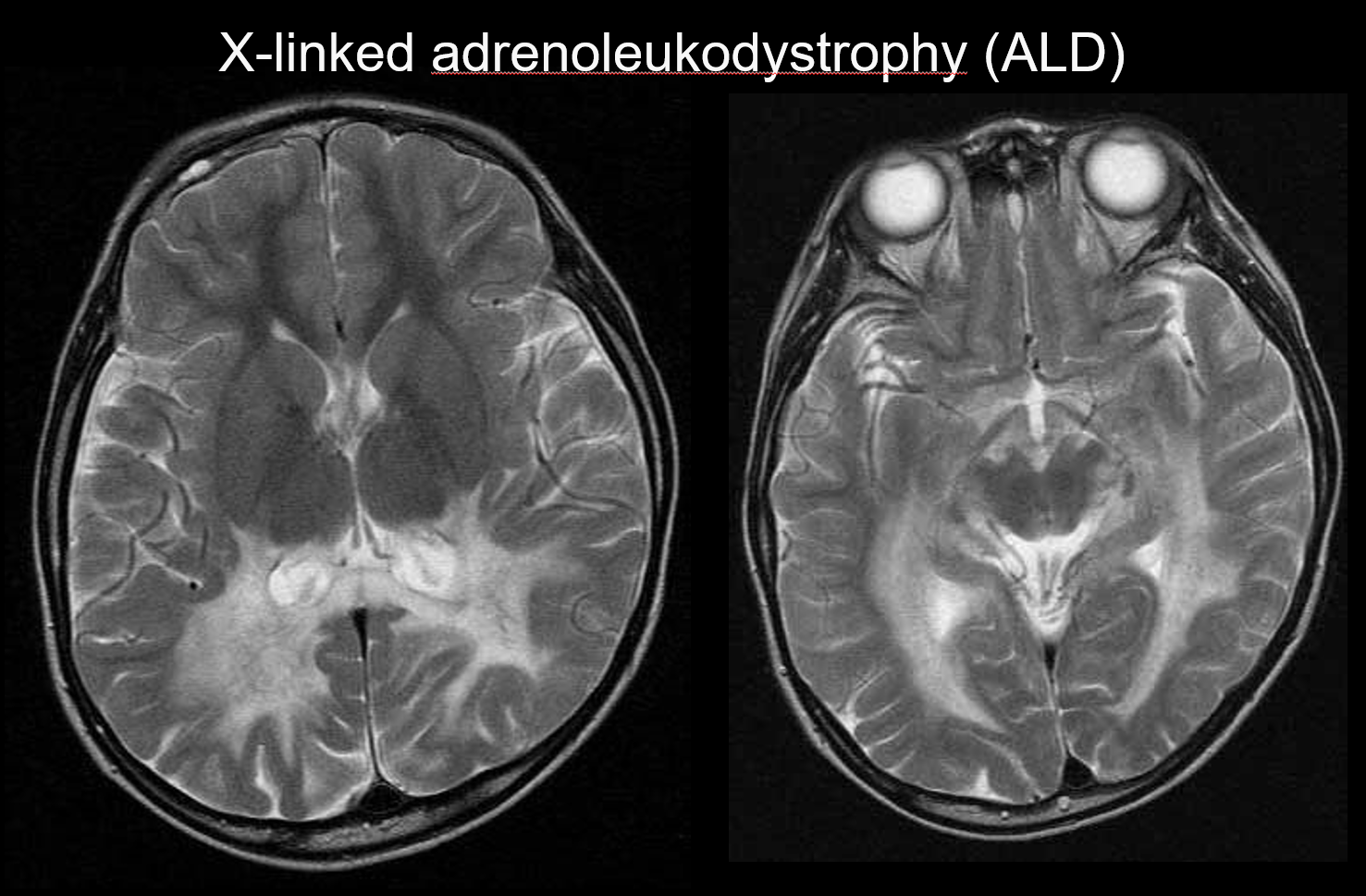
(b) Adrenoleukodystrofia sprzężona z chromosomem X
Adrenoleukodystrofia sprzężona z chromosomem X (ALD) jest recesywnym zaburzeniem dziedziczonym w sposób sprzężony z chromosomem X (zaburzenie peroksysomalne), spowodowanym nieprawidłowością genu ABCD1 (chromosom Xq28). Upośledzona β-oksydacja prowadzi do gromadzenia się bardzo długołańcuchowych kwasów tłuszczowych w istocie białej mózgu i nadnerczach, powodując demielinizację i niewydolność nadnerczy. ALD dzieli się na dziecięcą, młodzieńczą i dorosłą postać mózgową, adrenomieloneuropatię (AMN) oraz wyłącznie chorobę Addisona. Postać mózgowa dziecięca rozwija się w wieku 5-8 lat, z objawami obejmującymi pogorszenie sprawności intelektualnej, spastyczny chód, zaburzenia widzenia i słuchu. Patologicznie demielinizacja postępuje od istoty białej otaczającej trigon komory bocznej do splotu ciała modzelowatego, stopniowo rozszerzając się w kierunku przednio-bocznym. Odzwierciedlając patologię choroby, w MRI widoczne są symetryczne hiperintensywności T2 i hipointensywności T1 rozciągające się w kierunku przednio-bocznym od istoty białej otaczającej trójkąt komory bocznej, z widocznym wzmocnieniem kontrastowym na obrzeżach (ryc. 4). Widoczne są również zmiany w drogach korowo-rdzeniowych.
4. Przewaga okołokomorowa
Zaburzenia te charakteryzują się głównie zmianami w istocie białej otaczającej komory boczne, przy czym podkorowa istota biała (włókna U) jest zachowana. Taki wzorzec jest obserwowany w wielu zaburzeniach, w tym w MLD, i dlatego jest stosunkowo niespecyficzny. Łagodnie nieprawidłowe sygnały wokół komór bocznych są również widoczne w zwyrodnieniach korowych, szczególnie w neuronalnych lipofuscynozach ceroidalnych, które rozwijają się po okresie niemowlęcym.
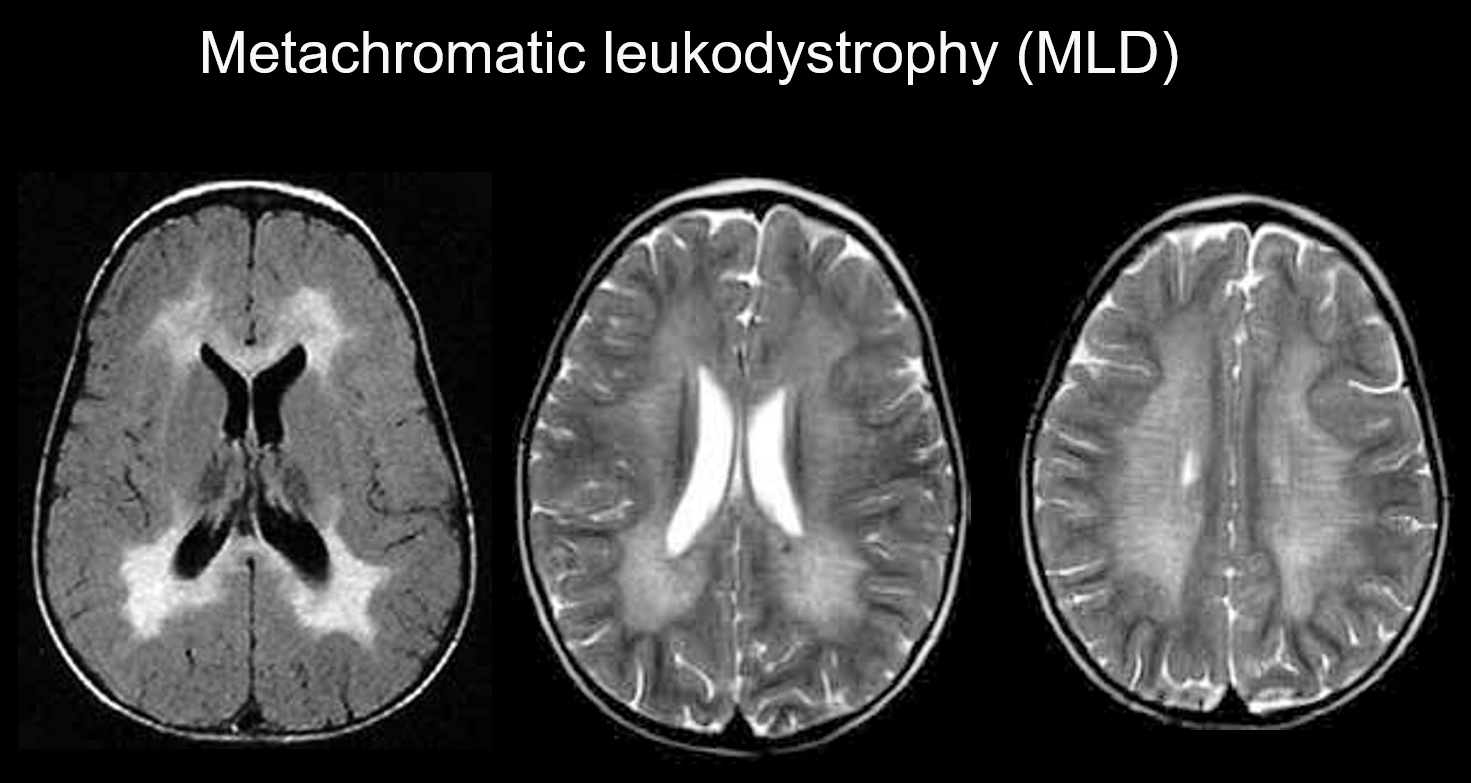
(a) Leukodystrofia metachromatyczna.
Leukodystrofia metachromatyczna jest dziedziczonym autosomalnie recesywnie zaburzeniem (lizosomalnym zaburzeniem spichrzeniowym) spowodowanym niedoborem arylosulfatazy-A (chromosom 22q13.31), w którym nagromadzenie wysoce toksycznego sulfatydu powoduje demielinizację. W zależności od wieku, w którym się pojawia, jest klasyfikowana jako wrodzona, o początku niemowlęcym, o początku młodzieńczym lub o początku dorosłym. Objawy obejmują regresję poznawczą, porażenie spastyczne, ruchy mimowolne, neuropatię obwodową i zanik nerwu wzrokowego. W obrazowaniu T2-ważonym objawia się jako hiperintensywność istoty białej, głównie wokół komór bocznych, a w obrazowaniu T1-ważonym jako łagodne hipointensywności. Zmiany występują głównie w płatach czołowych. W obrębie rozległych nieprawidłowych sygnałów w istocie białej mogą być widoczne pasma o prawidłowej intensywności (tygrysie prążki) (rysunek 5). Uważa się, że jest to spowodowane częściowym zachowaniem osłonki mielinowej w przestrzeni okołonaczyniowej i gromadzeniem się produktów rozpadu osłonki mielinowej w makrofagach.
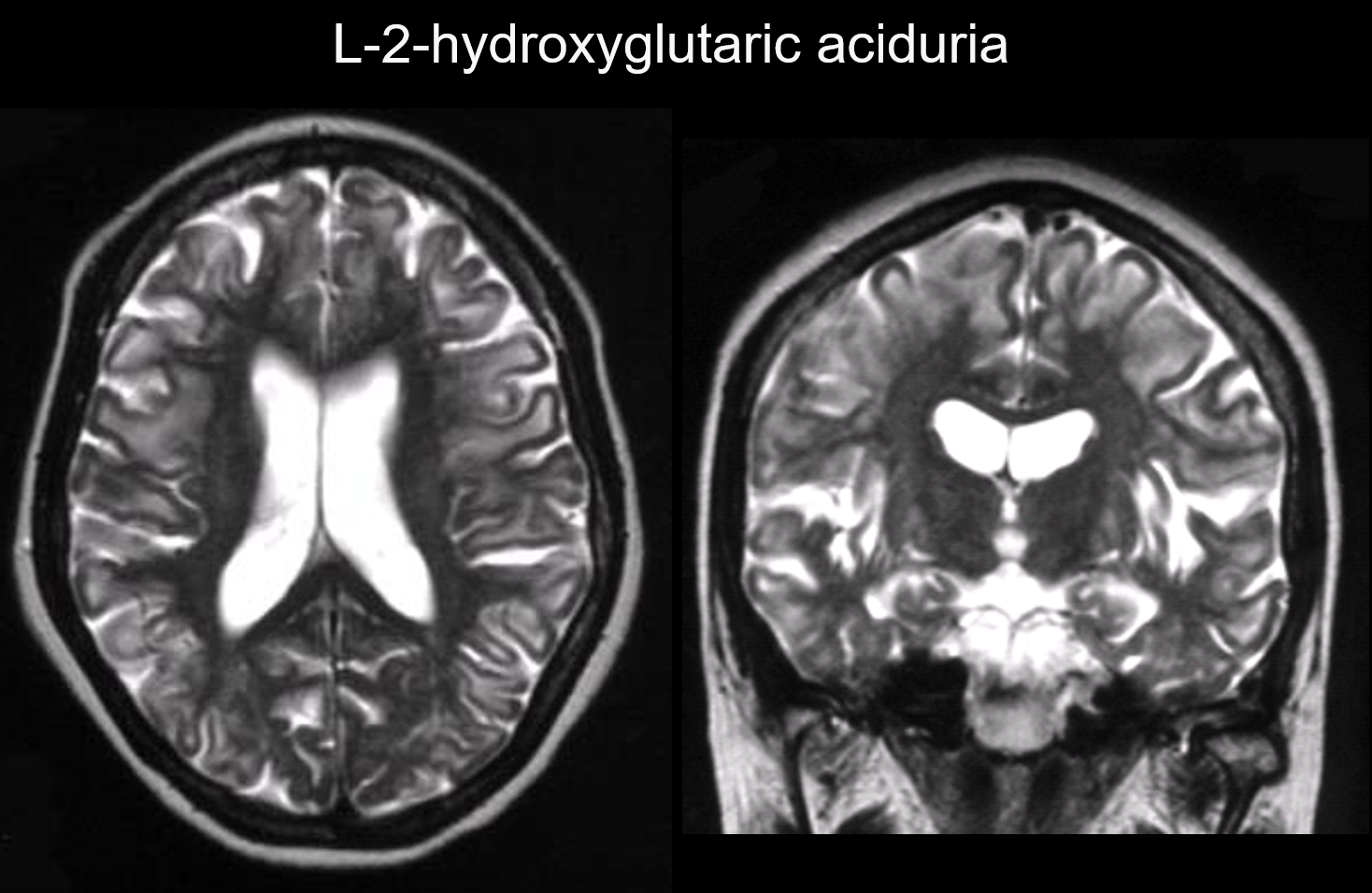
5. Dominacja podkorowa
W tych zaburzeniach zmiany występują głównie w podkorowej istocie białej, w tym we włóknach U. Zaburzenia o takim wzorcu obejmują kwasicę L-2-hydroksyglutarową (ryc. 6), galaktozemię, zespół Kearns-Sayer, akadamia propionowa, zaburzenia cyklu mocznikowego i wczesne stadium choroby Canavana.
6. Rozproszona istota mózgowa
W tych zaburzeniach nieprawidłowe sygnały pojawiają się w całej istocie białej mózgu. Wykazują one silne hiperintensywności T2 w porównaniu z sygnałami T2 wytwarzanymi przez niezmielinizowaną istotę białą (hipomielinizacja). Oprócz przypadków leukoencefalopatii megalencefalicznej z torbielami podkorowymi i leukoencefalopatii z zanikającą istotą białą, u pacjentów z każdym rodzajem zaburzeń istoty białej w miarę postępu choroby pojawia się ten wzorzec.
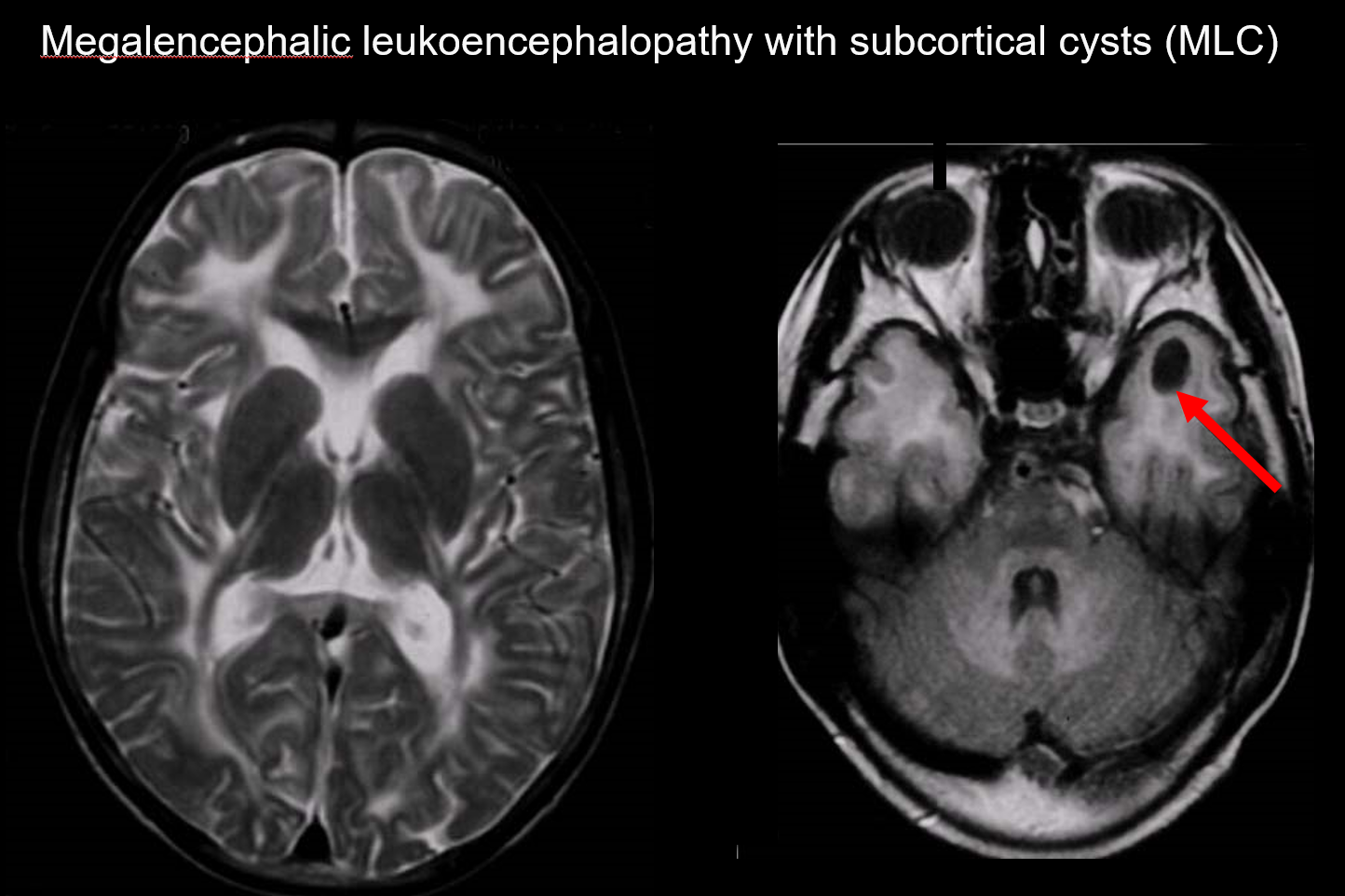
(a) Megalencefaliczna leukoencefalopatia z torbielami podkorowymi (MLC)
MLC jest dziedziczonym autosomalnie recesywnie zaburzeniem spowodowanym nieprawidłowością genu MLC1, a jej początek w okresie niemowlęcym charakteryzuje się megalocefalią, powoli postępującym pogorszeniem sprawności ruchowej, ataksją i spastycznością. MRI ujawnia charakterystyczne rozległe nieprawidłowe sygnały w istocie białej i łagodny obrzęk istoty białej, a także tworzenie torbieli w płatach ciemieniowych i skroniowych (ryc. 7).7, 8) Obrazowanie T1-zależne i T2-zależne ujawnia nieprawidłową istotę białą, podczas gdy torbiele wykazują hipointensywność T1 i hiperintensywność T2, co czyni je szczególnie trudnymi do wykrycia. Obrazowanie FLAIR, które uwidacznia torbiele (wodę) jako hipointensywności, jest cenne w diagnostyce. Wśród Japończyków występuje częściej niż wakuolizująca leukoencefalopatia megalencefaliczna.
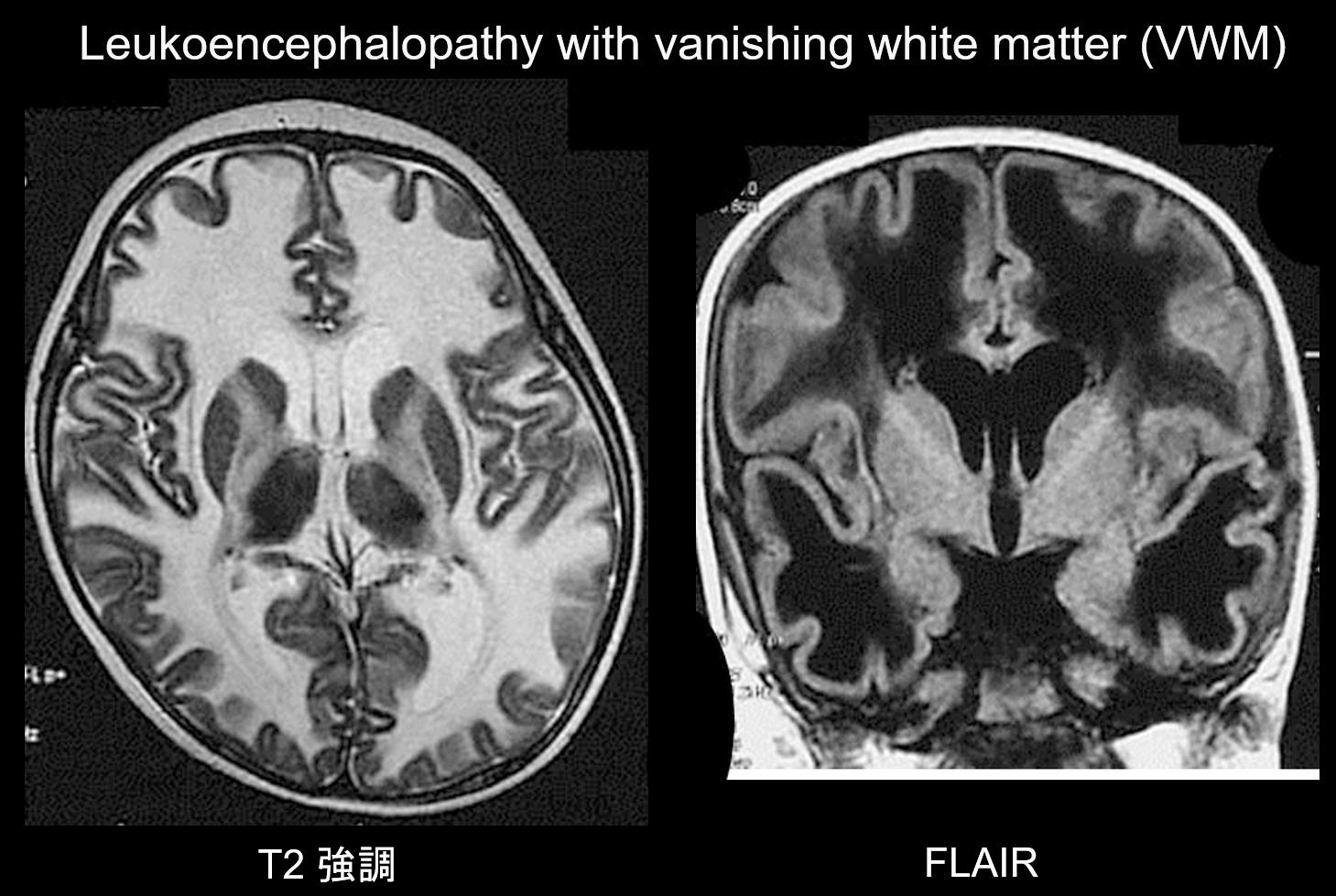
(b) Leukoencefalopatia ze zanikającą istotą białą.
Leukoencefalopatia ze zanikającą istotą białą (VWM) jest zaburzeniem dziedziczonym w sposób autosomalny recesywny, spowodowanym niedoborem eIF2B, białka związanego z eIF2, które przenosi inicjujący tRNA do rybosomów. eIF2B składa się z pięciu różnych białek, z których wszystkie mają różne loci genetyczne. Wykazano, że VWM jest tym samym zaburzeniem, co dziecięca ataksja móżdżkowa i centralna hipomielinizacja (CACH). Pacjenci są normalni w okresie noworodkowym i we wczesnym niemowlęctwie, ale po wystąpieniu choroby (zwykle w wieku 2-6 lat) rozwija się u nich powoli postępujący regres poznawczy, spastyczność i ataksja. Wiadomo, że objawy te nasilają się pod wpływem infekcji lub niewielkiego urazu. Istota biała mózgu wykazuje rozległą hiperintensywność T2 i hipointensywność T1 i z czasem jest stopniowo zastępowana przez płyn (jak sama nazwa wskazuje, istota biała zanika) (ryc. 8). Torbielowata istota biała zawiera pasmowate struktury, które uważa się za reprezentację pozostałej tkanki. Nieprawidłowe sygnały są również widoczne w pniu mózgu, szczególnie w centralnej części drogi okrężnej. Obrazowanie FLAIR jest cenne w diagnostyce tego zaburzenia.
7. Przewaga lub wybitność tylnego dołu czaszki
Zaburzenia te charakteryzują się zmianami dominującymi w pniu mózgu i móżdżku. Zmiany w istocie białej móżdżku mogą być spowodowane przez zaburzenia, w tym ksantomatozę mózgowo-rdzeniową (CTX), zaburzenia peroksysomalne, chorobę Alexandra, leukoencefalopatię z zajęciem pnia mózgu i rdzenia kręgowego oraz podwyższeniem stężenia mleczanów (LBSL), chorobę syropu klonowego, histiocytozę oraz toksyczność heroiny i kokainy. Zmiany w pniu mózgu mogą być spowodowane zaburzeniami takimi jak choroba Alexandra, LSBL i choroba poliglukozanowa dorosłych. Zmiany w środkowym odcinku szypuły móżdżku obserwuje się w zespole kruchego X i w autosomalnej dominującej leukodystrofii dorosłych związanej z duplikacją lamin B1.
8. Zmiany wieloogniskowe
W przeciwieństwie do zaburzeń, które powodują zmiany konfluentne opisane w punktach 2-7 powyżej, zaburzenia w tej sekcji powodują zmiany wieloogniskowe (rozproszone). Należą do nich infekcje, takie jak zespół TORCH (spowodowany wrodzonym zakażeniem cytomegalowirusem lub inną przyczyną) i bruceloza; zaburzenia zapalne, takie jak ostre rozsiane zapalenie mózgu i rdzenia kręgowego (ADEM), stwardnienie rozsiane (MS) i zapalenie nerwu wzrokowego (NMO); vasculopathies such as cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), atherosclerosis, amyloid angiopathy, COL4A1-associated cerebral small-vessel disease, Fabry disease, and Susac syndrome; oraz schorzenia dziedziczne, takie jak choroba mitochondrialna, kwasica L-2-hydroksyglutarowa, mukopolisacharydoza (MPS) i nieprawidłowości chromosomalne (takie jak zespół 6p).
9. Zmiany o niskiej zdolności dyfuzyjnej
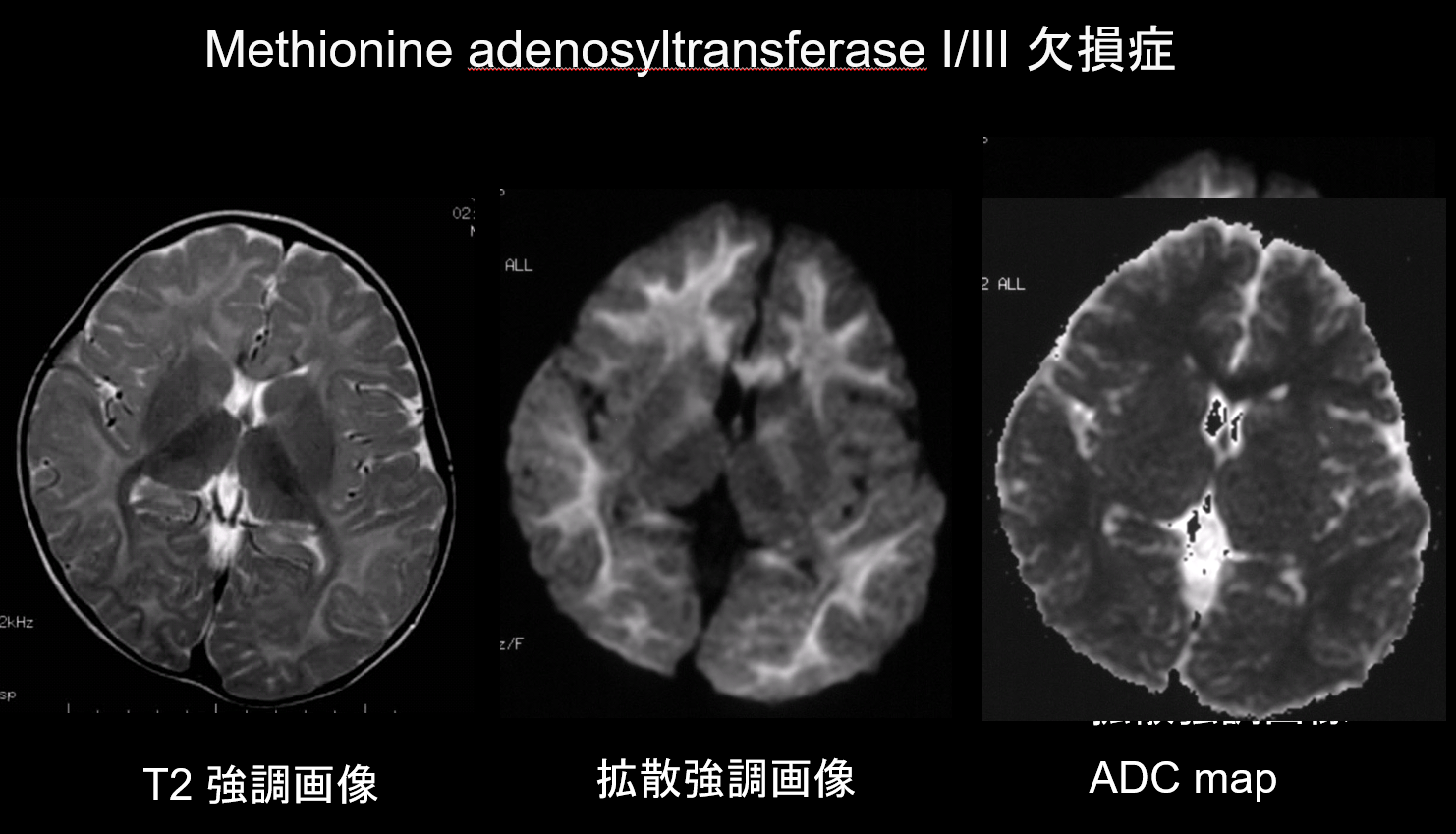
W zarówno demielinizacji, jak i hipomielinizacji, głównych patologiach zaburzeń istoty białej, dochodzi do zmniejszenia ilości mieliny, co ogranicza dyfuzję, a odpowiadający temu wzrost ilości płynu pozakomórkowego skutkuje hiperintensywnością T2 z wysokim pozornym współczynnikiem dyfuzji (ADC). Rzadko zdarza się, aby zaburzenia istoty białej wykazywały zarówno hiperintensywność T2, jak i niski ADC, dlatego taka kombinacja ma dużą wartość diagnostyczną. Zaburzenia charakteryzujące się obecnością obrzęku wewnątrzmielinowego w obrębie osłonek mielinowych i w szczelinach między osłonkami wykazują niski ADC. Należą do nich: choroba syropu klonowego, niedobór adenozylotransferazy metioninowej I/III (ryc. 9), fenyloketonuria, hiperglikemia nieketotyczna i choroba Canavana. Choroba Krabbego i leukodystrofia metachromatyczna mogą również wykazywać niskie ADC w niektórych zmianach w istocie białej, ponieważ obrzęk wewnątrzmielinowy może wystąpić podczas ostrej fazy demielinizacji.
- Van der Knaap MS, Valk J. Classification of myelin disorders. In Van der Knaap MS, Valk J, eds. Magnetic resonance of myelination and myelin disorders. 3rd ed. Berlin: Springer, 2005, 20-24.
- Schiffmann R, van der Knaap MS. An MRI-based approach to the diagnosis of white matter disorders. Neurology 2009; 72: 750-759
- Takanashi J. Diagnostic imaging of white matter disorders. Journal of the Japan Pediatric Society 2007; 111: 1243-1254.
- Van der Knaap MS, Breiter SN, Naidu S, et al. Defining and categorizing leukoencephalopathies of unknown origin: MR imaging approach. Radiology 1999; 213: 121-133.
.