Xa直接阻害剤の発見と開発
Factor Xa: 構造と結合部位編集

Xa因子の立体構造
Factor IIa、Xa、VIIa、IXa、XIaはいずれもタンパク質分解酵素で、凝固カスケードに特定の役割を担っている。 Xa因子は内因性経路と外因性経路の交差点に位置し、Xa1分子に対して約1000個のトロンビン分子を生成し、強力な抗凝固作用を示すことから、最も有望な因子とされている。 FXaは、52アミノ酸からなる活性化ペプチドが切断されることによりFXから生成されます(Xa因子の “a “は活性化を意味します)。 FXaは254アミノ酸の触媒ドメインからなり、さらに142アミノ酸の軽鎖が連結されている。
FXaの活性部位は生理的基質の切断を触媒する構造になっており、プロトロンビンのPhePheAsnProArg-ThrPheとTyrIleAspGlyArg-IleValを切断する。 FXaには4つのポケットがあり、これが基質がXa因子に結合する際のターゲットとなる。 これらのポケットは異なるアミノ酸によって並べられており、Xa阻害剤は第Xa因子に結合する際にこれらのポケットを標的とする。 S1:S1ポケットは疎水性ポケットで、塩基性基の認識部位となりうるアスパラギン酸残基(Asp-189)を含んでいる。 FXaはS1ポケットに残留空間を持ち,Tyr-228,Asp-189,Ser-195残基が並ぶ。
S2:S2ポケットは小さくて浅いポケットである。 S4ポケットと合流し、小さなアミノ酸のためのスペースを持っている。 Tyr-99はこのポケットへのアクセスをブロックしているようで,このポケットはS1やS4ほど重要ではない。
S3: S3ポケットはS1ポケットの縁に位置し,平らで溶媒にさらされている。 このポケットはS1やS4ほど重要ではない。
S4: S4ポケットはもともと疎水性で,ポケットの床はTrp-215残基によって形成されている. FXaのPhe-174とTyr-99残基はTrp-215と結合して芳香族ボックスを形成し,脂肪族,芳香族および正電荷の断片を結合することができる。 正電荷を持つ実体と結合するため、陽イオンホールと表現できる。
ダイレクトXa阻害剤の化学構造と特性編集
| Apixaban | Edoxaban | |||
|---|---|---|---|---|
| MW (g/mol) | 436 | 460 | 548 | |
| 分子量 formula | C19H18ClN3O5S | C25H25N5O4 | C24H30ClN7O4S | |
| 形状 | L | L | ||
| Ki | 0,08 nM | 0.561 nM | ||
| IC50 | 0.06 nM | N/A | ||
| 経口バイオアベイラビリティ(%) | 66-100 (用量-%) | N/A | ||
| 経口バイオアベイラビリティ(%) | N/A | 0.依存性) | 50 | 62 |

Xa阻害剤のXa因子への結合 Edit
Xa阻害剤はすべてXa因子の活性部位内でいわゆるL字型に結合する。 Xa因子の重要な構成要素はS1結合部位とS4結合部位である。 極性の高い、つまり帯電した成分を持つ天然化合物であるアンチスタシンやTAPが、ある特異性を持ってターゲットに結合することに初めて注目されたのである。 そのため、新しい薬剤は正電荷の基を持つように設計されたが、その結果、バイオアベイラビリティが悪くなった。 そこで、現在販売されているXa阻害剤は、S1およびS4結合部位と異なる相互作用をするために、さまざまな部位が結合した芳香環を含んでいます。 これにより、良好なバイオアベイラビリティが確保されるとともに、強固な結合強度が維持されるようになった。
Xa因子と結合するアンチスタシン
アンチスタシンはN末端とC末端のドメインを持ち、そのアミノ酸配列は約40%の同一性と約56%の相同性で類似しています。 それぞれ短いβ-シート構造と5つのジスルフィド結合を持つ。 Xaの阻害にはN末端ドメインのみが必要で、C末端ドメインは実際の活性部位に強く類似したパターンを持つにもかかわらず、3次元構造の違いから阻害特性には寄与しない。
アンチスタシンとFXaの相互作用には、FXaの活性部位と不活性表面の双方が関与している。 N-末端ドメインのArg-34とVal-35によって形成されるアンチスタシンの反応部位はFXaの結合部位、おそらくS1ポケットに適合している。 一方、アンチスタシンの反応部位の外側に位置するGlu-15は、FXa表面の正電荷を持つ残基と結合している。
DX-9065a binding to factor XaEdit
DX-9065a, the first small molecule direct Xa-inhibitor is a amidinoaryl derivate, with a molecular weight of 571.07g/mol. 正電荷を持つアミジノナフタレン基がFXaのS1ポケットにあるAsp-189残基と塩橋を形成している。
ヘパリンなどの旧来の薬剤とは異なり、DX-9065aはFXaとトロンビンの構造が似ているにもかかわらず、トロンビンと比較してFXaに選択的であることが特徴である。 これは、相同位置192のアミノ酸残基の違いによるものです。 FXaはその位置にグルタミン残基を持つのに対し、トロンビンはグルタミン酸を持ち、DX-9065aのカルボキシル基と静電反発を起こす。 さらに、トロンビンのGlu-97とDX-9065aのピロリジン環に固定されたアミジン基が塩橋をかけることで、DX-9065a分子の柔軟性が低下し、静電気の衝突を避けるために十分に回転することができなくなったのである。 そのため、トロンビンに対するIC50値は>1000μMであるのに対し、FXaに対するIC50値は0,16μMです。

Factor XaのポケットS1およびS4へのrivaroxabanの結合を示します。 2つの水素結合が形成され、リバーロキサバンをS1およびS4サブサイトへ導く重要な役割を担っている。 これらの水素結合により、リバーロキサバンはL字型を形成し、ポケットに収まる。 リバーロキサバンのS1ポケットのTyr-228と薬物の塩素置換基が相互作用し、リバーロキサバンが良好な経口バイオアベイラビリティと効力を達成することを可能にする
Rivaroxaban binding to factor XaEdit
FXaとの結合はアミノ酸Gly-219への2つの水素結合によって媒介されています。 これらの2つの水素結合は、FXaのS1およびS4サブサイトに薬物を誘導する重要な役割を担っている。 最初の水素結合は、リバーロキサバンのオキサゾリジノン骨格のカルボニル酸素に由来する強い相互作用である。 2つ目の水素結合は弱い相互作用で、クロロチオフェンカルボキサミド部分のアミノ基に由来します。
これらの2つの水素結合により、薬剤はL字型を形成し、S1およびS4ポケットに適合します。 Phe-174、Tyr-99、Trp-215のアミノ酸残基は、S4結合ポケットである狭い疎水性チャネルを形成しています。 リバーロキサバンのモルホリノン部分はアミノ酸Tyr-99とPhe-174の間に「挟まれて」おり、リバーロキサバンのアリール環はTrp-215を挟んで垂直な方向に向いている。
チオフェン部分の塩素置換基とS1の底に位置するTyr-228の芳香環との間の相互作用は、FXaに対する高い親和性のための強い塩基性基の必要性を排除するという事実によって、非常に重要である。 5599>

Apixaban binding to factor XaEdit
Apixaban is similar binding mode as arivaroxaban and form a tight inhibitor-enzyme complex when connected to FXa. アピキサバンのp-メトキシ基はFXaのS1ポケットに接続するが,FXaのこの領域のどの残基とも相互作用はないようである。 アピキサバンのピラゾールN-2窒素原子はGln-192と相互作用し、カルボニル酸素はGly-216と相互作用する。 アピキサバンのフェニルラクタム基はTyr-99とPhe-174の間に位置し、その配向性によりS4ポケットのTrp-215と相互作用することが可能である。 5599>

構造活性相関(SAR)編集
ある標的に対する理想的な阻害剤となる化合物を設計する上で重要なことは、化合物が結合する標的部位のアミノ酸配列について理解することである。 プロトロンビンとFXaの両方をモデル化することで、その違いを差し引き、それぞれの結合部位のアミノ酸を特定することが可能になります。 FXaのS1ポケットの底にある結合アミノ酸はAsp-189で、アミジン部分が結合することができる。 FXaの結合部位をX線で調べたところ、S1ポケットは平面的な形状をしており、平らなアミジノアリール基は立体障害なく結合できることがわかりました。 L字型の長辺は、標的の活性部位にある高度に特異的なトンネルに適合しなければならない。 そのために、分子のこの部分は、その領域でFXaとほとんど正式な相互作用を持たないように設計されている。 特異的な結合がないため、これらの薬剤がFXaのポケットの間に適合することで、FXa分子に対する薬剤の特異性が全体的に高まります。 FXaのS1ポケットと阻害剤の間の相互作用は、イオン性でも非イオン性でも可能であり、これは経口バイオアベイラビリティを高めるために部位の設計を調整することができるため重要である。 以前設計された化合物は荷電分子であり、消化管での吸収が悪いため、高い血清濃度に達しなかった。 新薬は帯電しておらず、S1ポケットに非イオン性の相互作用があるため、バイオアベイラビリティが向上しています。
リバロキサバンのSAR開発において、研究者はオキサゾリジンのコアに5-chlorothiophene-2-carboxamideグループを追加すると、これまで医療用途としては弱すぎた効力を200倍高めることに気づきました。 この発見に加え、(S)-配位への明確な嗜好性が確認された。 この化合物は、S1ポケットとの相互作用に重要であると考えられていた塩基性の高いアミジン基を含まず、薬物動態学的なプロファイルも有望であることが確認された。 これらの発見は、広範なSAR(構造活性相関)研究につながった。 SAR試験において、R1が効力に最も重要な基と定義された。 R1の官能基としては、ピロリジノンが最初に効力を大きく伸ばしましたが、さらなる研究の結果、代わりにモルホリノン基を用いると、さらに高い効力を発揮することがわかりました。 R2およびR3は、水素またはフッ素が結合した基であり、水素が結合していることが最も高い効力をもたらすとすぐに評価された。 そこで、R2、R3をいろいろな基と入れ替えてみたが、いずれも水素より効力が弱かったので、最終的に水素とした。 クロロチオフェン部分は水溶性が不十分であったため、他の基への置換を試みたが、うまくいかなかった。 クロロチオフェン部分は、S1ポケットの底にあるTyr-228に結合し、FXaとの結合に関する重要なファクターとなる。 リバーロキサバンは、高い親和性と良好なバイオアベイラビリティを併せ持つ。


SAR開発中、最大の効力とバイオアベイラビリティを達成するためにテストする必要があったグループが3つありました。 最初のグループは、p-メトキシフェニル基(S1結合部位)のSAR試験の前に安定化させる必要があるため、非活性部位を試験することになりました。 化合物の効力を高める基はいくつかあり、主にアミド、アミン、テトラゾールだが、メチルスルホニル基やトリフルオロメチル基もある。 これらの基のうち、カルボキサミドが最も結合力が強く、化合物と同様の凝固活性がありました。
犬の試験では、13Fというカルボキサミド基を持つこの化合物は、素晴らしい薬物動態プロファイルを示し、低いクリアランスと十分な半減期と分配容積を示しました。 安定化基の探索に成功したため、S1結合部位(p-methoxyphenyl)のSAR研究は中止された。 S4結合部では、N-メチルアセチルおよびラクタム類似体がFXaに対して非常に高い結合親和性を持ち、他のプロテアーゼに対して優れた凝集能と選択性を示すことが証明された。
SynthesisEdit
RivaroxabanEdit
リバロキサバンは化学的にはn-アリロキサゾリジノン系のグループに属している。 そのグループの他の薬剤はlinezolidとtedizolidであり、どちらも抗生物質である。 O-シリル保護されたエチル(2,3-ジヒドロキシプロピル)-カルバメートから始まるn-アリールオキサゾリジノン類の合成は2016年に発表された。 ワンポット反応により、カルバメートは弱塩基性条件下で2-オキサゾリドン環に環化し、同時にオキサゾリドン窒素は銅触媒によりアリール化されます。 特にリバーロキサバンでは、3-モルフォリノンが銅触媒作用によりベンゼン環のp位のヨウ素を置換する。 その後、シリル保護基を除去し、得られたアルコールをアミノ基で置換し、最後の工程でアシル化する。
2005年にバイエルヘルスケアによってリバーロキサバンの工業的製剤が特許として登録されている。 これはN-(4-アミノフェノール)-モルフォリノンから始まり、フタルイミド保護基に関与する第一級アミンをも含むプロピレンオキシド誘導体によってアルキル化される。 次に、ホスゲン当量を加えて2-オキサゾリドン環を形成し、フタルイミドを除去する。 5599>
しかし、特許によると、この合成は「準備に特に好ましくない影響を及ぼす反応管理におけるさまざまな欠点」を持っています。 この特許では、工業的なプロセスにもっと適したクロロチオフェン誘導体から始まる別の合成法も説明しているが、最終製品から有毒な溶媒や試薬を除去しなければならないことを指摘している。 したがって、この方法は代替案ではありません。
リバーロキサバンの様々な他の合成経路が記載されています。
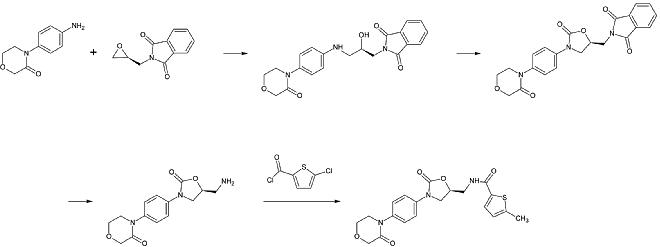
第1段。 第一級芳香族アミンのアルキル化
第二段階。 ホスゲン換算で2-オキサゾリニドン環を形成する
第3工程。 フタルイミド保護基の除去
第4工程。 第一級アミンのアシル化
ApixabanEdit
Apixabanの最初の完全合成は、2007年に発表された。 この反応の鍵となるステップは、p-methoxyphenylchlorohydrazon 誘導体と p-iodophenyl-morpholin-dihydropyridin 誘導体との (3+2)cycloaddition である。 続いて塩酸とモルホリンを除去した後、銅触媒によりヨウ素を2-ピペリジノンに置換し、エチルエステルからアミドに変換する(アミノリシス)。 この反応は2009年に特許登録されている。
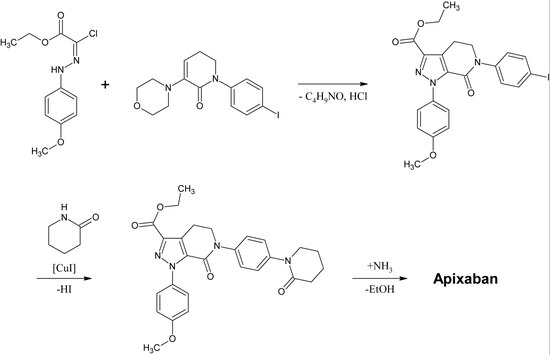
。