Imaging diagnostico delle anomalie della materia bianca | Ipomielinizzazione cerebrale congenita; rete per la malattia di Pelizaeus-Merzbacher e disturbi correlati
Immagini diagnostiche delle anomalie della materia bianca
Junichi TAKANASHI, Dipartimento di Pediatria, Tokyo Women’s Medical University, Yachiyo Medical Center
Introduzione
In questo articolo, descrivo l’approccio impiegato per la diagnosi dei disturbi che appaiono come segnali anormali nella materia bianca cerebrale sulla risonanza magnetica (MRI), dall’imaging alla diagnosi. I disturbi che colpiscono principalmente la materia bianca sono generalmente indicati come “leucoencefalopatia” o “disturbi della materia bianca” in inglese.1, 2) 3) Un altro termine, leucodistrofia, è a volte confuso con la degenerazione della materia bianca, ma questo in realtà si riferisce a uno spettro più ristretto di disturbi con una componente genetica (disturbi demielinizzanti ereditari).
Classificazione per immagini dei disturbi della materia bianca
L’avvento della risonanza magnetica ha notevolmente migliorato la nostra capacità di individuare le lesioni nella materia bianca del sistema nervoso centrale. Molte forme conosciute di disturbi della materia bianca mostrano segni specifici sulla risonanza magnetica, il che è utile per la loro diagnosi. Identificare i modelli di anormalità della materia bianca visti sulla risonanza magnetica (T1-pesata, T2-pesata o FLAIR imaging) rende facile restringere le possibilità in più diagnosi differenziali. La classificazione di Schiffmann e van der Kamp dei disturbi della materia bianca secondo i risultati della risonanza magnetica ha un valore pratico2, 4) (Figura 1, Tabella 1). Anche se questo non porta a una diagnosi finale, la classificazione dei risultati di imaging può portare alla successiva scoperta di un nuovo disturbo. Qui, descrivo i disturbi della materia bianca in termini di classificazioni MRI di cui sopra, e spiegare i principali tipi di disturbi.
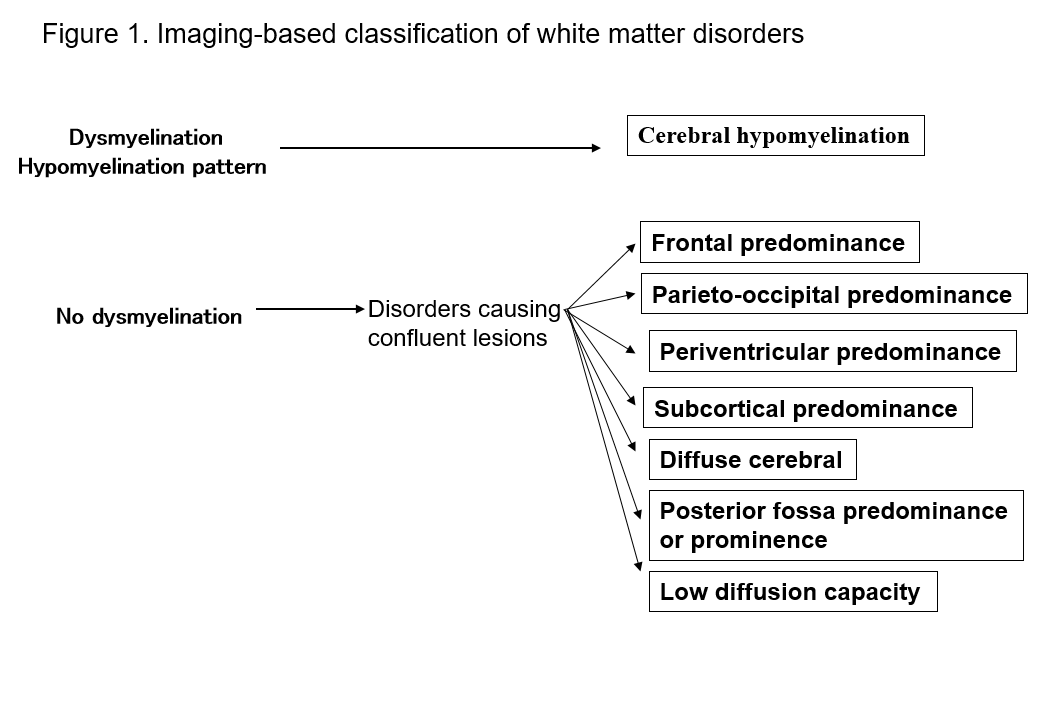
Tabella 1. Elenco di disturbi secondo i modelli di MRI
- Prevalenza frontale
Malattia di Alexander, variante frontale dell’adrenoleucodistrofia X-linked (ALD), leucodistrofia metacromatica (MLD), leucodistrofia neuroassonale con sferoidi. - Predominanza parieto-occipitale
Adrenoleucodistrofia X-linked (ALD), malattia di Krabbe, disturbi perossisomiali ad insorgenza precoce, ipoglicemia neonatale. - Prevalenza periventricolare
Leucodistrofia metacromatica (MLD), malattia di Krabbe, sindrome di Sjögren-Larsson, malattia dei corpi poliglucanici adulti, leucoencefalopatia con coinvolgimento del tronco encefalico e del midollo spinale ed elevazione del lattato (LBSL), leucomalacia periventricolare (PVL), encefalopatia da HIV, lipofuscinosi ceroide neuronale ad insorgenza tardiva. - Predominanza sottocorticale
L-2-idrossiglutarico aciduria, galattosemia, sindrome di Kearns-Sayer, accademia propionica, disturbi del ciclo dell’urea, malattia di Canavan. - Leucoencefalopatia cerebrale diffusa
Megalencefalica con cisti sottocorticali (MLC), leucoencefalopatia con materia bianca scomparsa (VWM), distrofia muscolare congenita carente di merosina, malattia mitocondriale, carenza di cofattore di molibdeno, carenza di solfito ossidasi, casi avanzati di disturbi della materia bianca. - Prevalenza o prominenza della fossa posteriore
Lesioni del cervelletto e dei peduncoli cerebellari: xantomatosi cerebrotendinea (CTX), disturbi perossisomiali, malattia di Alexander, leucoencefalopatia con coinvolgimento del tronco cerebrale e del midollo spinale ed elevazione del lattato (LBSL), malattia delle urine da sciroppo d’acero, istiocitosi, leucodistrofia autosomica dominante adulta legata ad una duplicazione della lamina B1, tossicità da eroina e cocaina.
Lesioni del tronco cerebrale: Malattia di Alexander, LSBL, disturbi perossisomiali, malattia di Wilson, malattia poliglucosica adulta, sindrome di Leigh, atrofia dentatorubropallidoluysiana (DRPLA), malattia del corpo poliglucosico adulto, leucodistrofia autosomica dominante adulta legata ad una duplicazione della lamina B1. - Lesioni multifocali
Sindrome di Torch (infezione congenita da citomegalovirus), brucellosi, encefalomielite acuta disseminata (ADEM), sclerosi multipla (SM), neuromielite ottica (NMO), arteriopatia cerebrale autosomica dominante con infarti subcorticali e leucoencefalopatia (CADASIL), aterosclerosi, angiopatia amiloide, malattia dei piccoli vasi cerebrali associata a COL4A1, malattia di Fabry, sindrome di Susac, malattia mitocondriale, aciduria L-2-idrossiglutarica, mucopolisaccaridosi (MPS), anomalie cromosomiche (come la sindrome 6p). - Lesioni con bassa capacità di diffusione
Malattia delle urine da sciroppo d’acero, deficit di metionina adenosiltransferasi I/III, fenilchetonuria, iperglicinemia non chetotica, malattia di Canavan, lesioni attive nella malattia di Krabbe e leucodistrofia metacromatica.
1. Ipomielinizzazione della materia bianca cerebrale
Si riferisce a un gruppo di disturbi in cui la formazione della guaina mielinica è compromessa o ritardata, e le sue immagini assomigliano a quelle dei neonati con mielinizzazione immatura. Nelle immagini pesate in T2, la materia bianca appare caratteristicamente come una diffusa iperintensità che è debole rispetto alla corteccia. Per maggiori dettagli, vedere il sito web dell’ipomielinizzazione cerebrale congenita.
Se le lesioni della materia bianca non sono coerenti con l’ipomielinizzazione della materia bianca cerebrale, si deve determinare se sono confluenti o multiple.2) Le lesioni confluenti della materia bianca sono di solito dovute alla degenerazione ereditaria della materia bianca (leucodistrofia) e nella maggior parte dei casi sono bilateralmente simmetriche. Le lesioni multiple della materia bianca sono di solito asimmetriche e acquisite. Le lesioni confluenti della materia bianca sono ulteriormente suddivise nelle categorie 2-7 seguenti.
2. Predominanza frontale
In questo gruppo di disturbi, estese lesioni della materia bianca sono presenti prevalentemente nel lobo frontale. Includono la malattia di Alexander, la variante frontale dell’adrenoleucodistrofia X-linked (ALD), la leucodistrofia metacromatica (MLD) e la leucodistrofia neuroassonale con sferoidi.
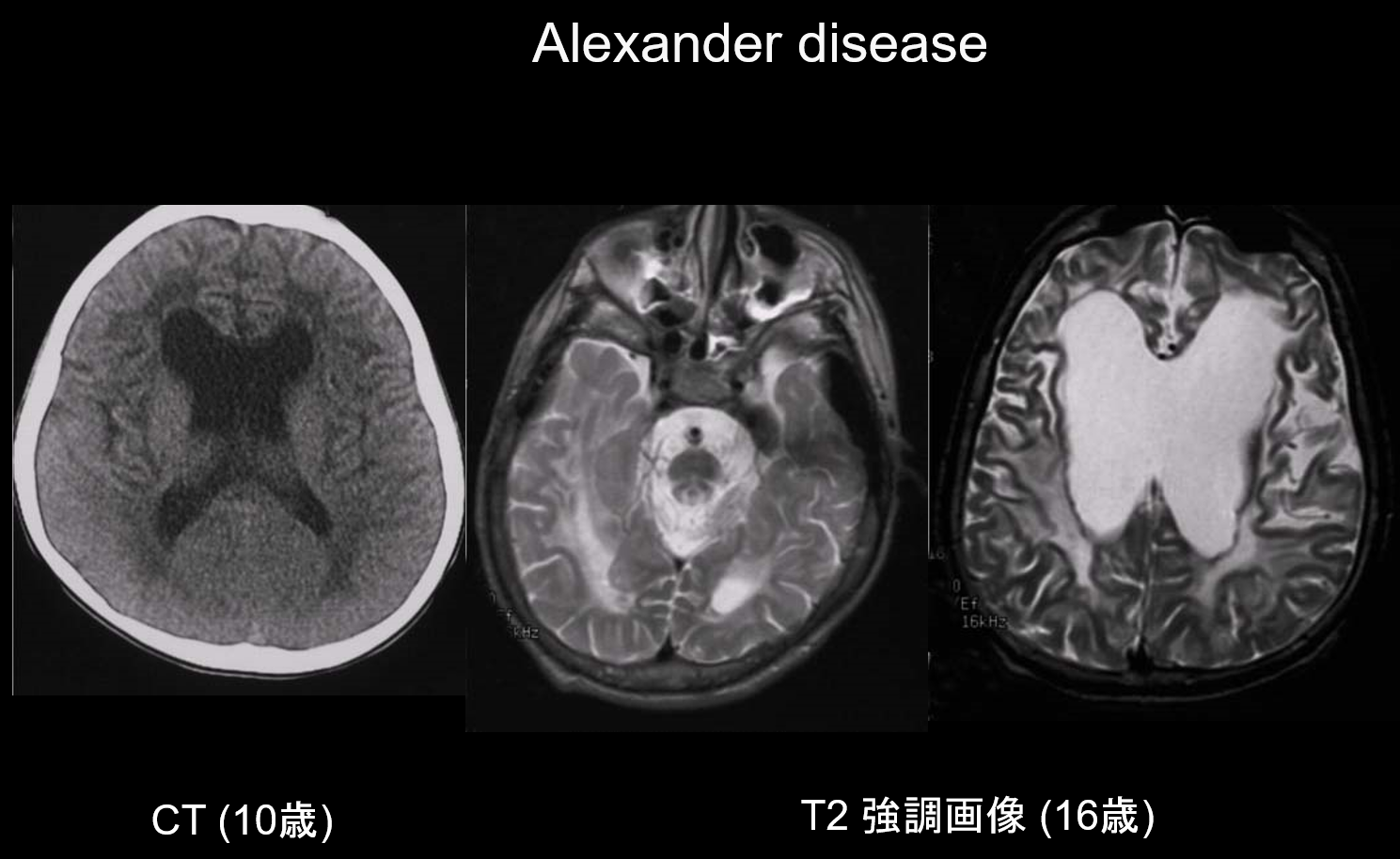
(a) La malattia di Alexander.
La malattia di Alexander è un disturbo ereditario autosomico dominante causato da una mutazione nel gene GFAP sul cromosoma 17q21. Risulta nell’accumulo di fibre di Rosenthal nelle cellule gliali stellate. Queste fibre sono composte da GFAP e proteine dello stress (αB-cristallina e HSP27). La malattia di Alexander si manifesta principalmente nell’infanzia, tra i 3 mesi e i 2 anni, con la comparsa di megalencefalia, ritardo dello sviluppo, paralisi spastica ed epilessia. Alla risonanza magnetica, può presentare (i) lesioni diffuse della materia bianca, prevalentemente nel lobo frontale; (ii) marginazione T1 iperintensa e T2 ipointensa intorno ai ventricoli laterali; (iii) lesioni nei gangli della base e nel talamo; (iv) lesioni del tronco encefalico; e (v) aumento del contrasto delle lesioni attive (Figura 2). Nelle fasi iniziali, insieme alle lesioni della materia bianca e del putamen, si vede un gonfiore che può gradualmente causare atrofia o formazione di cisti.
3. Predominanza parieto-occipitale
La caratteristica principale di questo gruppo di disturbi sono le lesioni della materia bianca parieto-occipitale. Includono l’adrenoleucodistrofia X-linked (ALD), la malattia di Krabbe, i disturbi perossisomiali ad insorgenza precoce e l’ipoglicemia neonatale.
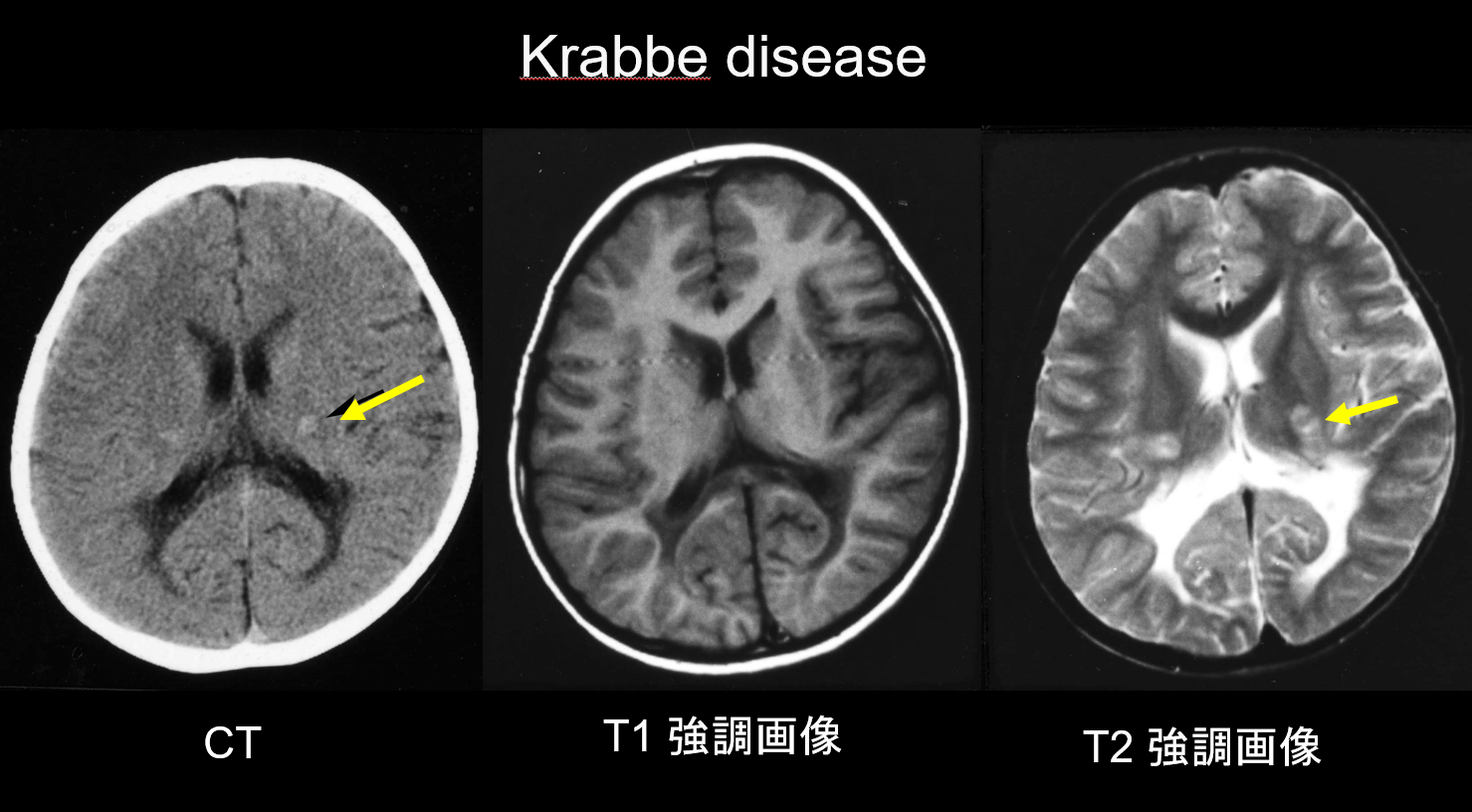
(a) Malattia di Krabbe.
La malattia di Krabbe è un disordine ereditario autosomico recessivo (malattia da accumulo lisosomiale) causato dalla carenza di galattosilceramidasi (cromosoma 14q31), in cui l’accumulo di psicosina altamente citotossica si ritiene causi una demielinizzazione diffusa. Compaiono anche grandi cellule multinucleate chiamate “cellule globoidi”. A seconda dell’età in cui compare, viene classificata come malattia infantile, infantile a esordio tardivo, a esordio giovanile o a esordio adulto. La maggior parte dei casi sono infantili e iniziano con la comparsa di febbre, irritabilità, difficoltà di alimentazione, ritardo nello sviluppo, neuropatia periferica, spasticità e atrofia del nervo ottico all’età di 3-6 mesi. Durante le prime fasi, la tomografia computerizzata (TC) rivela una caratteristica iperdensità nel talamo e nella corona radiata. Si ritiene che questo rifletta le cellule globoidi ad alta densità e la proliferazione gliale. La risonanza magnetica può anche mostrare iperintensità T1 e ipointensità T2 intorno ai ventricoli, così come strutture lineari simili a quelle viste nella MLD (Figura 3). Il nucleo dentato cerebellare, la materia bianca cerebellare e il tratto piramidale del tronco encefalico mostrano iperintensità T2 da uno stadio iniziale.
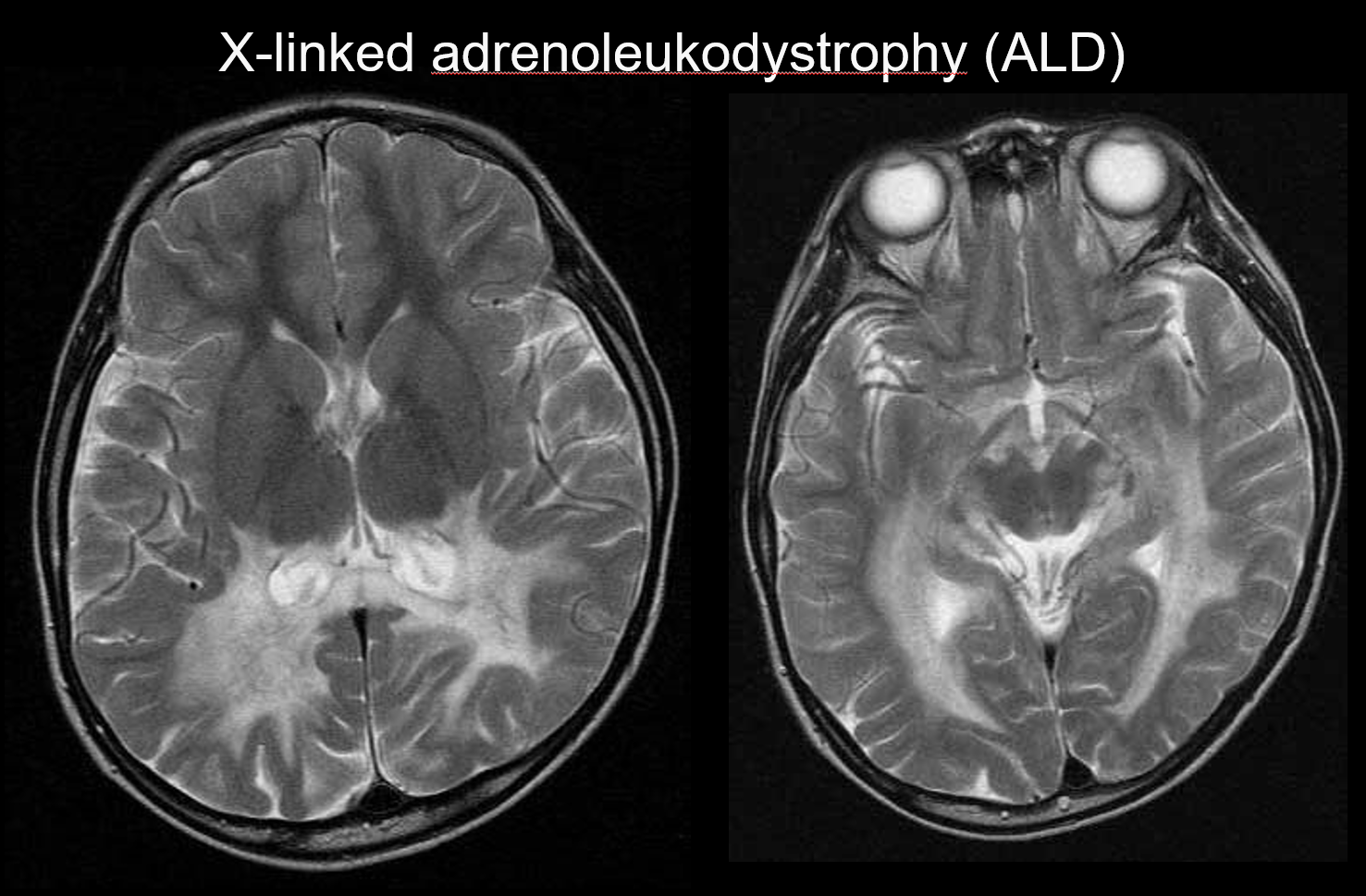
(b) Adrenoleucodistrofia legata all’X
L’adrenoleucodistrofia legata all’X (ALD) è un disordine ereditario recessivo legato all’X (disturbo perossisomiale) causato da un’anomalia del gene ABCD1 (cromosoma Xq28). L’alterazione della β-ossidazione porta all’accumulo di acidi grassi a catena molto lunga nella materia bianca cerebrale e nelle ghiandole surrenali, causando demielinizzazione e insufficienza surrenale. L’ALD è classificata nelle forme cerebrali infantile, adolescenziale e adulta, nell’adrenomieloneuropatia (AMN) e nella sola malattia di Addison. La forma cerebrale infantile si sviluppa a 5-8 anni di età, con la comparsa di sintomi tra cui il deterioramento intellettuale, l’andatura spastica, la visione e l’udito compromessi. Patologicamente, la demielinizzazione progredisce dalla materia bianca che circonda il trigono del ventricolo laterale allo splenio del corpo calloso, estendendosi gradualmente anterolateralmente. Riflettendo la patologia della malattia, iperintensità simmetriche T2 e ipointensità T1 che si estendono anterolateralmente dalla materia bianca che circonda il trigono del ventricolo laterale sono evidenti su MRI, con aumento di contrasto evidente ai margini (Figura 4). Lesioni del tratto corticospinale sono anche evidenti.
4. Predominanza periventricolare
Questi disturbi sono caratterizzati principalmente da lesioni nella materia bianca che circonda i ventricoli laterali, con la materia bianca sottocorticale (fibre a U) conservata. Questo modello è visto in numerosi disturbi, tra cui MLD, ed è quindi relativamente non specifico. Segnali leggermente anormali intorno ai ventricoli laterali si vedono anche nella degenerazione corticale, in particolare nella lipofuscinosi ceroide neuronale che si sviluppa dopo l’infanzia.
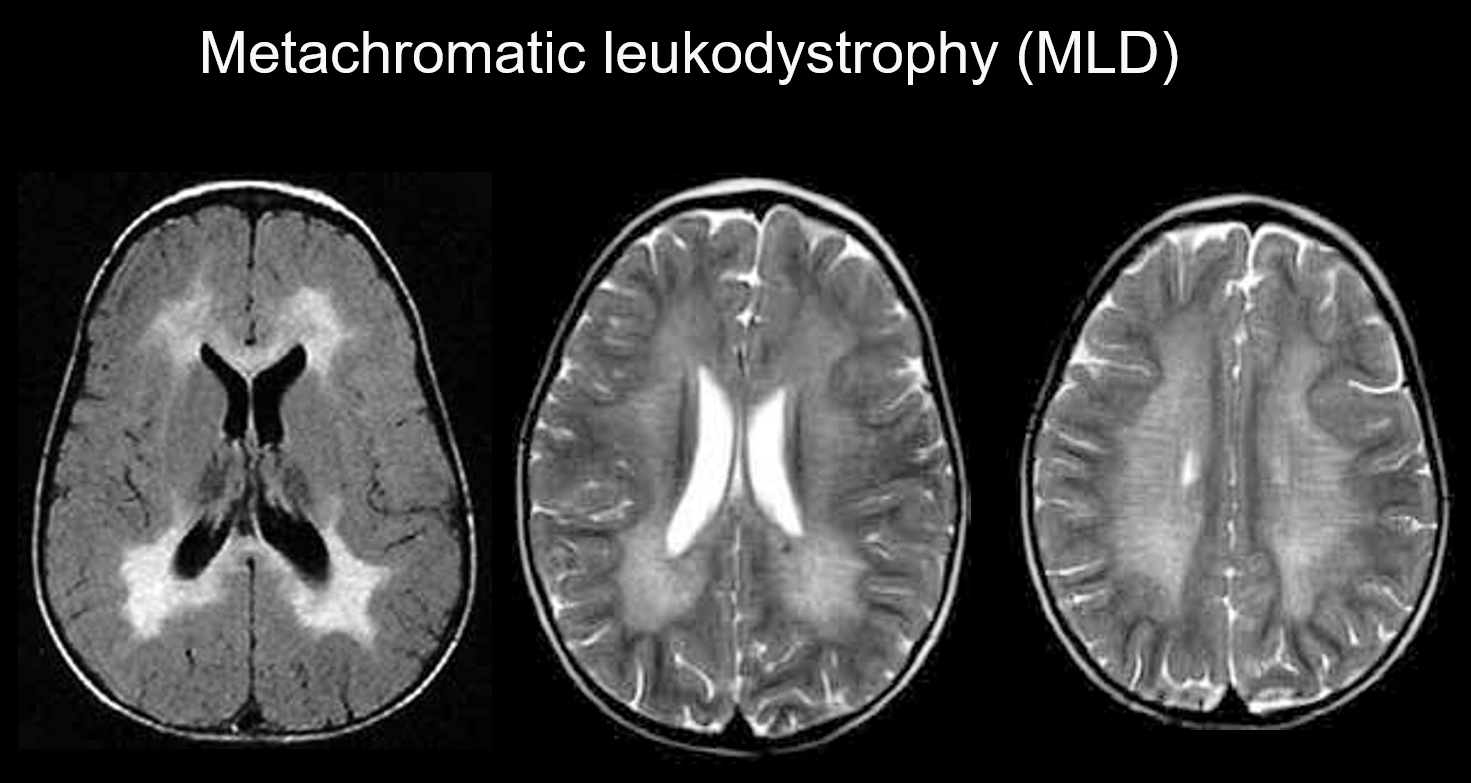
(a) Leucodistrofia metacromatica.
La leucodistrofia metacromatica è un disordine ereditario autosomico recessivo (disordine da accumulo lisosomiale) causato dalla carenza di arilsolfatasi-A (cromosoma 22q13.31), in cui l’accumulo di solfatidi altamente tossici provoca la demielinizzazione. A seconda dell’età in cui compare, viene classificata come congenita, a insorgenza infantile, giovanile o adulta. I suoi sintomi includono regressione cognitiva, paralisi spastica, movimenti involontari, neuropatia periferica e atrofia del nervo ottico. Appare sull’imaging ponderato T2 come iperintensità della materia bianca principalmente intorno ai ventricoli laterali, e sull’imaging ponderato T1 come lieve ipointensità. Le lesioni tendono ad essere prevalentemente nel lobo frontale. Bande di intensità normale (strisce di tigre) possono essere evidenti all’interno dei segnali anormali diffusi nella materia bianca (Figura 5). Si ritiene che queste siano dovute alla parziale conservazione della guaina mielinica nello spazio perivascolare e all’accumulo di prodotti di degradazione della guaina mielinica nei macrofagi.
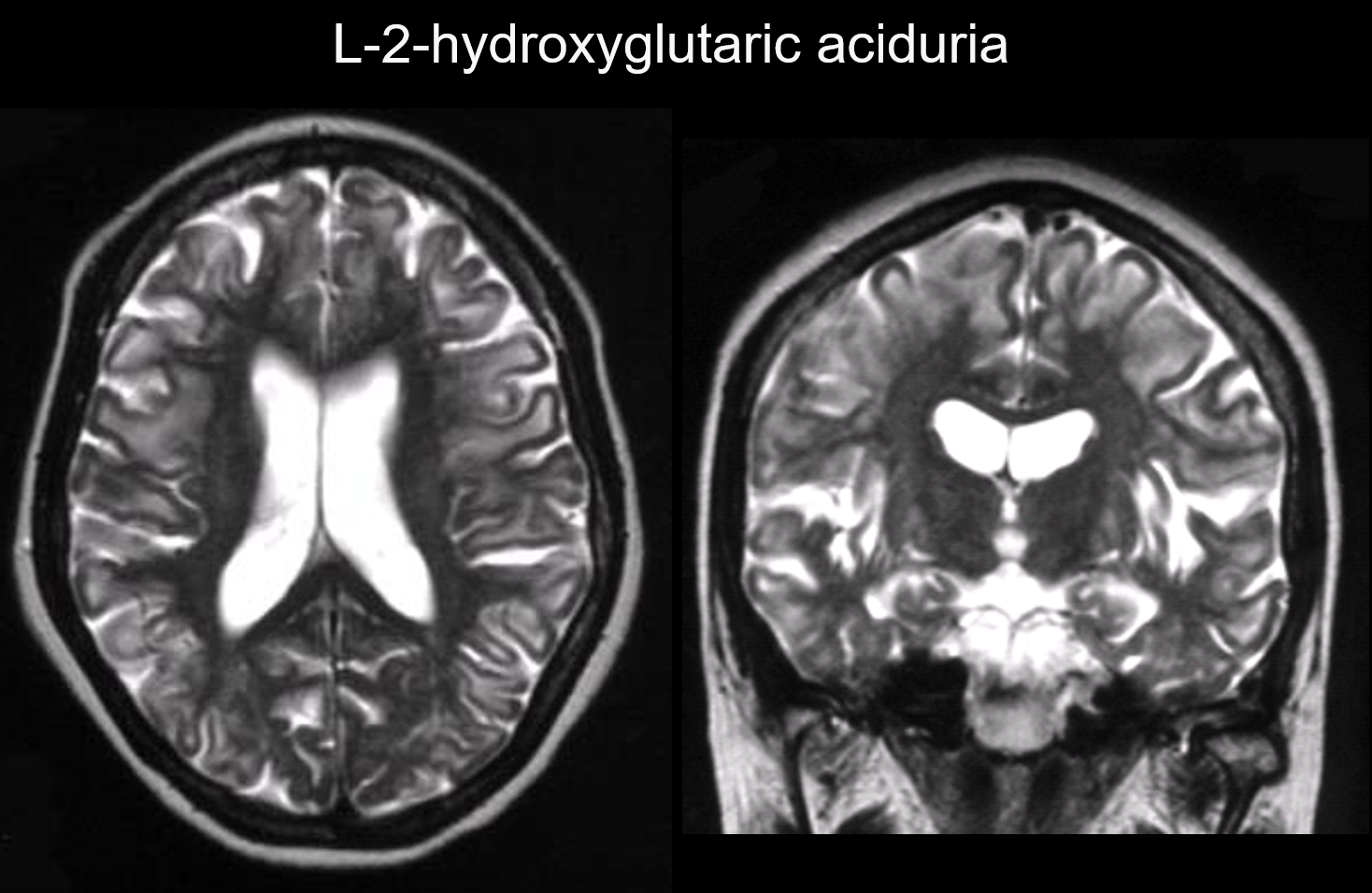
5. Predominanza sottocorticale
In questi disturbi, le lesioni si verificano principalmente nella materia bianca sottocorticale, comprese le fibre U. I disordini con questo modello includono l’aciduria L-2-idrossiglutarica (Figura 6), la galattosemia, la sindrome di Kearns-Sayer, l’accademia propionica, i disturbi del ciclo dell’urea e la malattia di Canavan allo stadio iniziale.
6. Cerebrale diffusa
In questi disturbi, segnali anormali appaiono in tutta la materia bianca cerebrale. Presentano forti iperintensità T2 rispetto ai segnali T2 prodotti dalla materia bianca non mielinizzata (ipomielinizzazione). Oltre ai casi di leucoencefalopatia megalencefalica con cisti sottocorticali e leucoencefalopatia con materia bianca scomparsa, i pazienti con qualsiasi tipo di disturbo della materia bianca alla fine mostrano questo modello con l’avanzare della malattia.
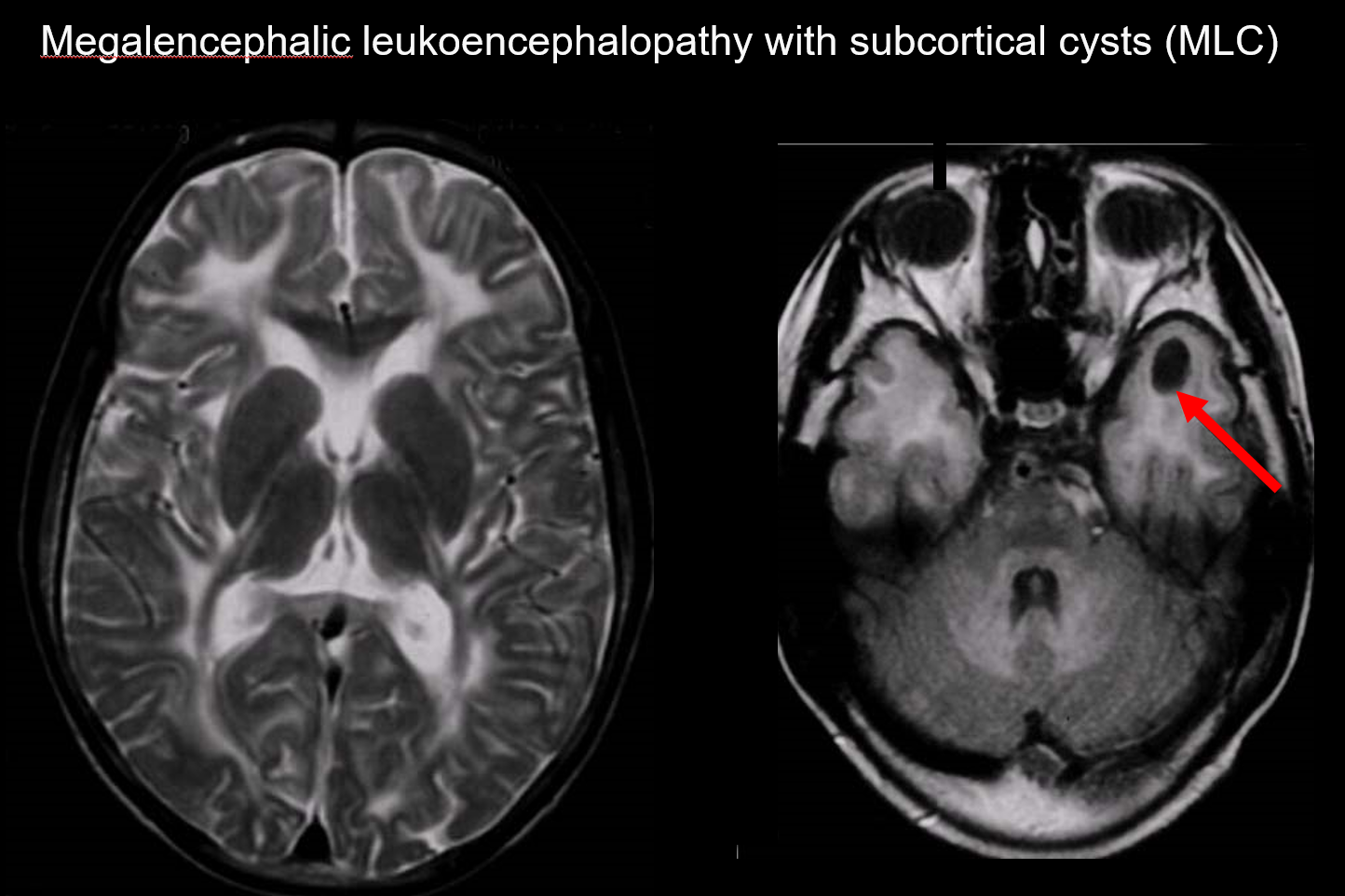
(a) Leucoencefalopatia megalencefalica con cisti sottocorticali (MLC)
MLC è un disordine ereditario autosomico recessivo causato da un’anomalia del gene MLC1, e il suo esordio nell’infanzia è caratterizzato da megalocefalia, deterioramento motorio che procede lentamente, atassia e spasticità. La risonanza magnetica rivela caratteristici segnali anormali diffusi nella materia bianca e un lieve rigonfiamento della materia bianca, nonché la formazione di cisti nei lobi parietali e temporali (Figura 7).7, 8) L’imaging T1-pesato e T2-pesato rivela materia bianca anormale, mentre le cisti presentano tutte ipointensità T1 e iperintensità T2, rendendole particolarmente difficili da individuare. L’imaging FLAIR, che visualizza le cisti (acqua) come ipointensità, è prezioso per la sua diagnosi. È più comune tra i giapponesi rispetto alla leucoencefalopatia megalencefalica vacuolante.
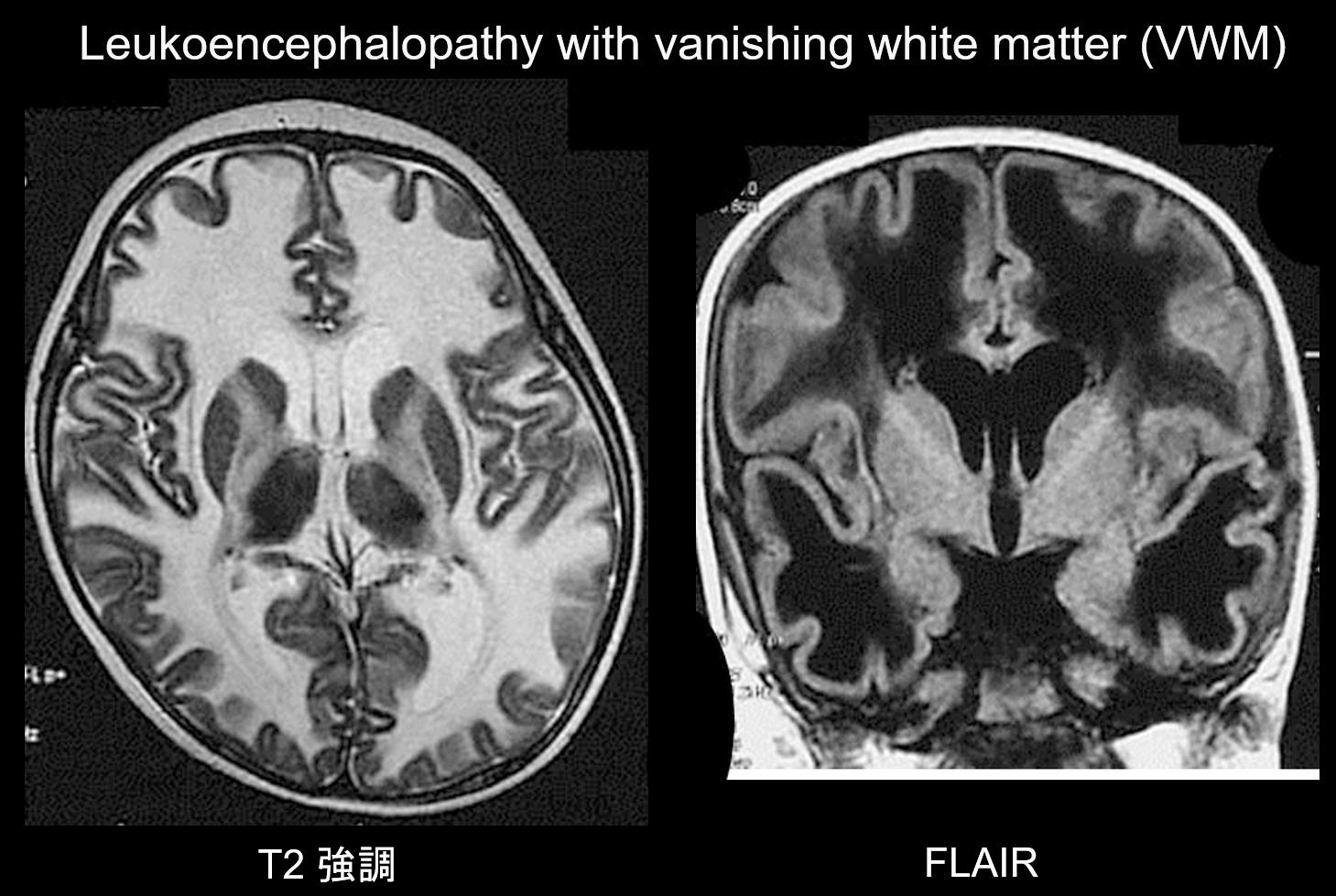
(b) Leucoencefalopatia con materia bianca scomparsa.
La leucoencefalopatia con materia bianca scomparsa (VWM) è un disordine ereditario autosomico recessivo causato da una carenza di eIF2B, una proteina associata a eIF2, che trasferisce il tRNA iniziatore ai ribosomi. eIF2B consiste di cinque diverse proteine, che hanno tutte diversi loci genetici. È stato dimostrato che VWM è lo stesso disordine dell’atassia cerebellare infantile e dell’ipomielinizzazione centrale (CACH). I pazienti sono normali durante il periodo neonatale e la prima infanzia, ma dopo l’esordio (di solito all’età di 2-6 anni) sviluppano una regressione cognitiva lentamente progressiva, spasticità e atassia. Questi sintomi sono noti per essere esacerbati da infezioni o traumi minori. La materia bianca cerebrale mostra una diffusa iperintensità T2 e ipointensità T1 ed è gradualmente sostituita da fluido nel tempo (come implica il nome, la materia bianca scompare) (Figura 8). La materia bianca cistica contiene strutture a bande che si ritiene rappresentino il tessuto rimanente. Segnali anomali si vedono anche nel tronco encefalico, in particolare nel tratto tegmentale centrale. L’imaging FLAIR è prezioso per la diagnosi di questo disturbo.
7. Predominanza o prominenza della fossa posteriore
Questi disturbi sono caratterizzati da lesioni prevalentemente nel tronco encefalico e nel cervelletto. Le lesioni della materia bianca cerebellare possono essere causate da disturbi come la xantomatosi cerebrotendinea (CTX), i disturbi perossisomiali, la malattia di Alexander, la leucoencefalopatia con coinvolgimento del tronco cerebrale e del midollo spinale ed elevazione del lattato (LBSL), la malattia dello sciroppo d’acero, l’istiocitosi e la tossicità da eroina e cocaina. Le lesioni del tronco cerebrale possono essere causate da disturbi come la malattia di Alexander, la LSBL e la malattia del poliglucosio adulto. Le lesioni del peduncolo cerebellare medio si vedono nella sindrome della X fragile e nella leucodistrofia autosomica dominante dell’adulto legata ad una duplicazione della lamina B1.
8. Lesioni multifocali
A differenza dei disturbi che producono le lesioni confluenti descritte in 2-7 sopra, i disturbi in questa sezione risultano in lesioni multifocali (sparse). Includono infezioni come la sindrome TORCH (dovuta a infezione congenita da citomegalovirus o altra causa) e la brucellosi; disturbi infiammatori come l’encefalomielite acuta disseminata (ADEM), la sclerosi multipla (MS) e la neuromielite ottica (NMO); vasculopatie come l’arteriopatia cerebrale autosomica dominante con infarti sottocorticali e leucoencefalopatia (CADASIL), aterosclerosi, angiopatia amiloide, malattia dei piccoli vasi cerebrali associata a COL4A1, malattia di Fabry e sindrome di Susac; e condizioni ereditarie come la malattia mitocondriale, l’aciduria L-2-idrossiglutarica, la mucopolisaccaridosi (MPS) e le anomalie cromosomiche (come la sindrome 6p).
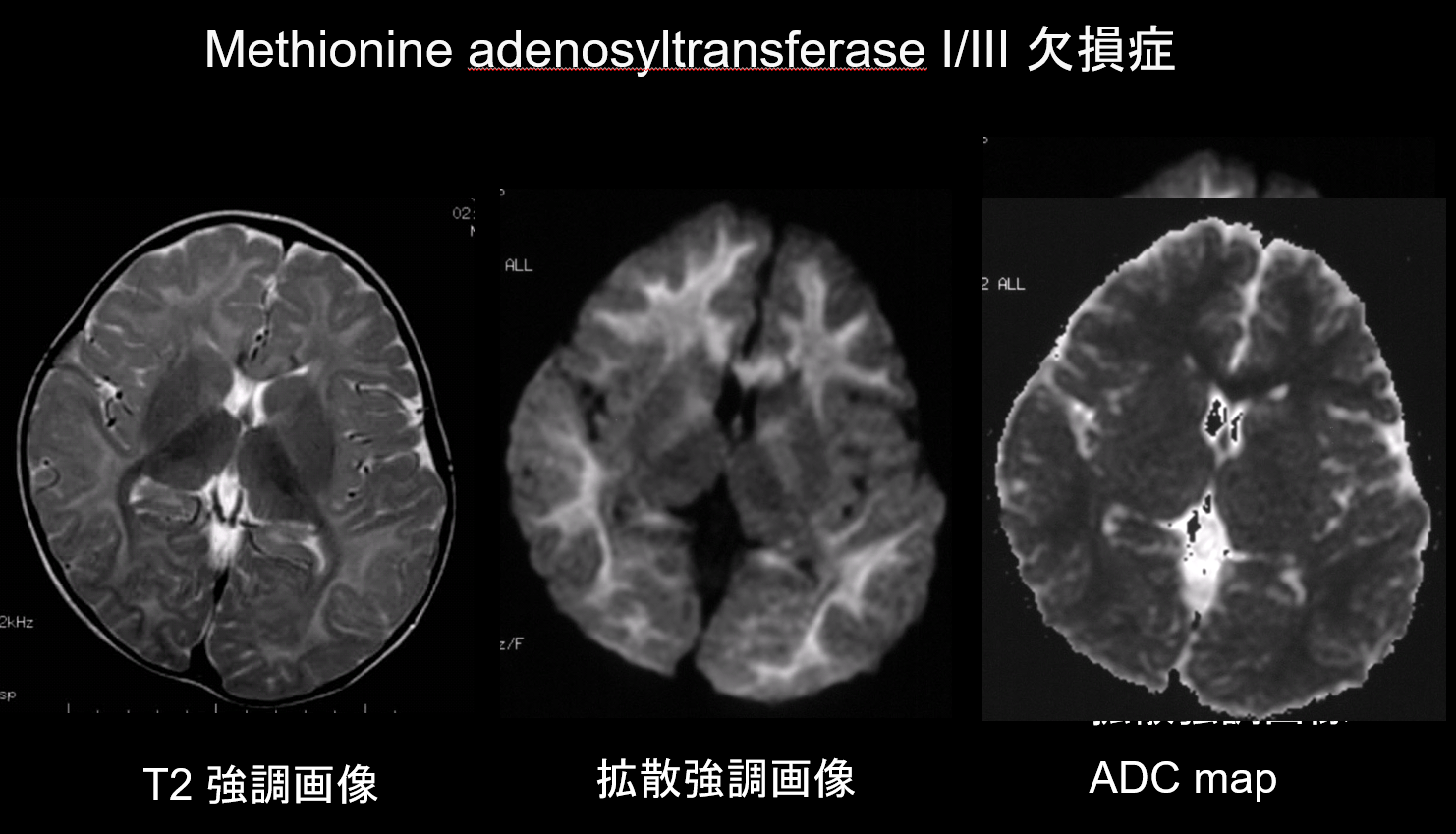
9. Lesioni con bassa capacità di diffusione
In entrambe le demielinizzazioni e ipomielinizzazioni, le principali patologie dei disturbi della materia bianca, c’è una diminuzione della quantità di mielina, che limita la diffusione, e il corrispondente aumento del fluido extracellulare provoca iperintensità T2 con un alto coefficiente di diffusione apparente (ADC). È raro che i disturbi della materia bianca presentino sia iperintensità T2 che un basso ADC, e questa combinazione ha quindi un alto valore diagnostico. Disturbi caratterizzati dalla presenza di edema intramielinico all’interno della guaina mielinica e negli spazi tra le guaine mostrano un ADC basso. Essi comprendono la malattia dello sciroppo d’acero nelle urine, il deficit di metionina adenosiltransferasi I/III (Figura 9), la fenilchetonuria, l’iperglicinemia non chetotica e la malattia di Canavan. La malattia di Krabbe e la leucodistrofia metacromatica possono anche mostrare una bassa ADC in alcune lesioni della materia bianca, come edema intramielinico può verificarsi durante la fase acuta della demielinizzazione.
- Van der Knaap MS, Valk J. Classificazione dei disturbi della mielina. In Van der Knaap MS, Valk J, eds. Risonanza magnetica della mielinizzazione e disturbi della mielina. 3rd ed. Berlino: Springer, 2005, 20-24.
- Schiffmann R, van der Knaap MS. Un approccio basato sulla risonanza magnetica per la diagnosi dei disturbi della materia bianca. Neurology 2009; 72: 750-759
- Takanashi J. Diagnostica per immagini dei disturbi della materia bianca. Journal of the Japan Pediatric Society 2007; 111: 1243-1254.
- Van der Knaap MS, Breiter SN, Naidu S, et al. Definizione e categorizzazione di leucoencefalopatie di origine sconosciuta: MR approccio di imaging. Radiologia 1999; 213: 121-133.