Découverte et développement d’inhibiteurs directs du Xa
Facteur Xa : Structure et sites de liaisonEdit

Les facteurs IIa, Xa, VIIa, IXa et XIa sont tous des enzymes protéolytiques qui ont un rôle spécifique dans la cascade de la coagulation. Le facteur Xa (FXa) est le plus prometteur en raison de sa position à l’intersection des voies intrinsèque et extrinsèque ainsi que de sa capacité à générer environ 1000 molécules de thrombine pour chaque molécule de Xa, ce qui entraîne un puissant effet anticoagulant. Le FXa est généré à partir du FX par clivage d’un peptide d’activation de 52 acides aminés, le « a » du facteur Xa signifiant activé. Le FXa est constitué d’un domaine catalytique de 254 acides aminés et est également lié à une chaîne légère de 142 acides aminés. La chaîne contient à la fois un domaine GLA et deux domaines de facteur de croissance épidermique (domaines semblables à l’EGF).
Le site actif du FXa est structuré pour catalyser le clivage de substrats physiologiques et clive PhePheAsnProArg-ThrPhe et TyrIleAspGlyArg-IleVal dans la prothrombine. Le FXa possède quatre « poches » qui sont des cibles pour les substrats qui se lient au facteur Xa. Ces poches sont alignées par différents acides aminés et les inhibiteurs du facteur Xa ciblent ces poches lorsqu’ils se lient au facteur Xa. Les deux poches les plus pertinentes en ce qui concerne l’affinité et la sélectivité pour les inhibiteurs du Xa sont S1 et S4.
S1 : La poche S1 est une poche hydrophobe et contient un résidu d’acide aspartique (Asp-189) qui peut servir de site de reconnaissance pour un groupe basique. FXa a un espace résiduel dans la poche S1 et est bordé par les résidus Tyr-228, Asp-189 et Ser-195.
S2 : La poche S2 est une poche petite et peu profonde. Elle fusionne avec la poche S4 et a de la place pour de petits acides aminés. Tyr-99 semble bloquer l’accès à cette poche, donc cette poche n’est pas aussi importante que S1 et S4.
S3 : La poche S3 est située sur le bord de la poche S1 et est plate et exposée au solvant. Cette poche n’est pas aussi importante que S1 et S4.
S4 : La poche S4 est de nature hydrophobe et le plancher de la poche est formé par le résidu Trp-215. Les résidus Phe-174 et Tyr-99 du FXa rejoignent Trp-215 pour former une boîte aromatique capable de lier des fragments aliphatiques, aromatiques et chargés positivement. En raison de la liaison à des entités chargées positivement, il peut être décrit comme un trou cationique.
Structure chimique et propriétés des inhibiteurs directs du XaEdit
| Rivaroxaban | Apixaban | Edoxaban | |
|---|---|---|---|
| MW (g/mol) | 436 | 460 | 548 |
| Moléculaire formule | C19H18ClN3O5S | C25H25N5O4 | C24H30ClN7O4S |
| Forme | L | L | L |
| Ki | 0.4 nM | 0,08 nM | 0,561 nM |
| IC50 | 0.7 nM | N/A | N/A |
| Biodisponibilité orale (%) | 66-100 (dose-dépendante) | 50 | 62 |

La liaison des inhibiteurs du Xa au facteur XaEdit
Les inhibiteurs du Xa se lient tous de manière dite en forme de L dans le site actif du facteur Xa. Les constituants clés du facteur Xa sont les sites de liaison S1 et S4. On a d’abord remarqué que les composés naturels, l’antistasin et le TAP, qui possèdent des composants hautement polaires et donc chargés, se lient à la cible avec une certaine spécificité. C’est pourquoi des médicaments plus récents ont été conçus avec des groupes chargés positivement, mais leur biodisponibilité était faible. Les inhibiteurs de la Xa commercialisés aujourd’hui contiennent donc un noyau aromatique auquel sont attachés plusieurs fragments pour différentes interactions avec les sites de liaison S1 et S4. Cela permet également d’assurer une bonne biodisponibilité tout en maintenant une force de liaison solide. Les inhibiteurs de Xa actuellement sur le marché, s’appuient donc sur des liaisons hydrophobes et hydrogène au lieu d’interactions hautement polaires.
La liaison de l’antistasin au facteur XaEdit
L’antistasin contient un domaine N- et un domaine C-terminal qui sont similaires dans leurs séquences d’acides aminés avec ~40% d’identité et ~56% d’homologie. Chacun d’entre eux contient une courte structure β-feuille et 5 liaisons disulfure. Seul le domaine N-terminal est nécessaire pour inhiber Xa tandis que le domaine C-terminal ne contribue pas aux propriétés inhibitrices en raison des différences dans la structure tridimensionnelle, même si le domaine C-terminal a un modèle fortement analogue au site actif réel.
L’interaction de l’antistasine avec le FXa implique à la fois le site actif et la surface inactive du FXa. Le site réactif de l’antistasine formé par Arg-34 et Val-35 dans le domaine N-terminal convient au site de liaison du FXa, très probablement la poche S1. Dans le même temps, Glu-15 situé à l’extérieur du site réactif de l’antistase correspond à des résidus chargés positivement sur la surface du FXa. La liaison multiple est thermodynamiquement avantageuse et conduit à une inhibition sub-nanomolaire (Ki = 0,3-0,6 nM).
La liaison de DX-9065a au facteur XaEdit
DX-9065a, la première petite molécule inhibitrice directe de Xa, est un dérivé amidinoaryle avec un poids moléculaire de 571,07g/mol. Son groupe amidinonaphtalène chargé positivement forme un pont salin avec le résidu Asp-189 dans la poche S1 du FXa. Le cycle pyrrolidine s’insère entre Tyr-99, Phe-174 et Trp-215 dans la poche S4 du FXa.
Contrairement aux médicaments plus anciens, par exemple l’héparine, le DX-9065a est sélectif pour le FXa par rapport à la thrombine, même si le FXa et la thrombine sont similaires dans leur structure. Ceci est dû à une différence dans le résidu d’acide aminé en position 192 de l’homologue. Alors que le FXa possède un résidu glutamine à cette position, la thrombine possède un acide glutamique qui provoque une répulsion électrostatique avec le groupe carboxyle du DX-9065a. De plus, un pont salin entre le Glu-97 de la thrombine et le groupe amidine fixé dans le cycle pyrrolidine du DX-9065a réduit la flexibilité de la molécule de DX-9065a, qui ne peut plus tourner suffisamment pour éviter le choc électrostatique. C’est pourquoi la valeur de la CI50 pour la thrombine est >1000µM alors que la valeur de la CI50 pour le FXa est de 0,16µM.

La liaison du rivaroxaban au facteur XaEdit
La liaison du rivaroxaban au FXa est médiée par deux liaisons hydrogène à l’acide aminé Gly-219. Ces deux liaisons hydrogène jouent un rôle important en dirigeant le médicament vers les sous-sites S1 et S4 du FXa. La première liaison hydrogène est une interaction forte qui provient de l’oxygène du carbonyle du noyau oxazolidinone du rivaroxaban. La seconde liaison hydrogène est une interaction plus faible et provient du groupe amino de la partie carboxamide du clorothiophène.
Ces deux liaisons hydrogène font que le médicament forme une forme en L et s’insère dans les poches S1 et S4. Les résidus d’acides aminés Phe-174, Tyr-99 et Trp-215 forment un canal hydrophobe étroit qui constitue la poche de liaison S4. La partie morpholinone du rivaroxaban est » prise en sandwich » entre les acides aminés Tyr-99 et Phe-174 et le cycle aryle du rivaroxaban est orienté perpendiculairement à travers Trp-215. Le groupe carbonyle de la morpholinone n’a pas d’interaction directe avec le squelette du FXa, au lieu de cela, il contribue à une planarisation de l’anneau de morpholinone et soutient donc le rivaroxaban pour être pris en sandwich entre les deux acides aminés.
L’interaction entre le substituant de chlore de la fraction thiophène et l’anneau aromatique de Tyr-228, qui est situé au bas du S1, il est très important en raison du fait qu’il évite le besoin de groupes fortement basiques pour une haute affinité pour le FXa. Cela permet au rivaroxaban, qui n’est pas basique, d’obtenir une bonne biodisponibilité et une bonne puissance orales.

La liaison de l’apixaban au facteur XaEdit
L’apixaban présente un mode de liaison similaire à celui du rivaroxaban et forme un complexe inhibiteur-enzyme étanche lorsqu’il est connecté au FXa. Le groupe p-méthoxy de l’apixaban se connecte à la poche S1 du FXa mais ne semble pas avoir d’interaction avec les résidus de cette région du FXa. L’atome d’azote N-2 du pyrazole de l’apixaban interagit avec Gln-192 et l’oxygène du carbonyle interagit avec Gly-216. Le groupe phényl lactame de l’apixaban est positionné entre Tyr-99 et Phe-174 et, en raison de son orientation, il est capable d’interagir avec Trp-215 de la poche S4. Le groupe oxygène carbonyle du fragment lactame interagit avec une molécule d’eau et ne semble interagir avec aucun résidu de la poche S4.

Structure-activité-relation (SAR)Edit
Une partie importante de la conception d’un composé, qui est un inhibiteur idéal pour une certaine cible, est de comprendre la séquence d’acides aminés du site cible pour que le composé se lie. La modélisation de la prothrombine et du FXa permet de déduire la différence et d’identifier les acides aminés de chaque site de liaison. Au fond de la poche S1 sur le FXa, l’acide aminé de liaison est l’Asp-189, auquel des fragments d’amidine peuvent se lier. Après avoir radiographié le site de liaison du FXa, il a été révélé que la poche S1 avait une forme plane, ce qui signifie qu’un groupe amidinoaryle plat devrait s’y lier sans encombrement stérique.
Les inhibiteurs directs modernes du Xa sont des molécules en forme de L dont les extrémités s’insèrent parfaitement dans les poches S1 et S4. Le côté long de la forme en L doit se conformer à un tunnel très spécifique dans le site actif des cibles. Pour y parvenir, cette partie des molécules est conçue pour avoir peu d’interactions formelles avec le FXa dans cette région. Comme il n’y a pas de liaison spécifique, l’ajustement de ces agents entre les poches du FXa augmente la spécificité totale des médicaments à la molécule de FXa. L’interaction entre la poche S1 du FXa et l’inhibiteur peut être à la fois ionique et non ionique, ce qui est important car cela permet d’ajuster la conception de la fraction pour augmenter la biodisponibilité orale. Les composés conçus précédemment étaient des molécules chargées qui ne sont pas bien absorbées dans le tractus gastro-intestinal et n’atteignent donc pas des concentrations sériques élevées. Les nouveaux médicaments ont une meilleure biodisponibilité car ils ne sont pas chargés et ont une interaction non ionique avec la poche S1.
Rivaroxaban
Pendant le développement SAR du rivaroxaban, les chercheurs ont réalisé que l’ajout d’un groupe 5-chlorothiophène-2-carboxamide au noyau oxazolidonine pouvait multiplier par 200 la puissance, qui était auparavant trop faible pour une utilisation médicale. En plus de cette découverte, une nette préférence pour la configuration (S) a été confirmée. Ce composé avait un profil pharmacocinétique prometteur et ne contenait pas de groupe amidine hautement basique, mais qui avait auparavant été considéré comme important pour l’interaction avec la poche S1. Ces résultats ont conduit à des recherches approfondies sur la relation structure-activité (SAR). Au cours des tests SAR, R1 a été défini comme le groupe le plus important pour la puissance. La pyrrolidinone a été le premier groupe fonctionnel R1 à augmenter significativement la puissance, mais des recherches ultérieures ont révélé une puissance encore plus élevée avec un groupe morpholinone à la place. Les groupes R2 et R3 étaient liés à l’hydrogène ou au fluor et il a été rapidement déterminé que l’hydrogène donnait la puissance la plus élevée. Les groupes R2 et R3 ont ensuite été remplacés par divers groupes, qui étaient tous moins puissants que l’hydrogène, de sorte que l’hydrogène a été le résultat final. La solubilité dans l’eau de la fraction chlorothiophène étant inadéquate, on a tenté de la remplacer par un autre groupe, mais sans succès. Le groupement chlorothiophène se lie à Tyr-228 au fond de la poche S1, ce qui en fait un facteur clé de la liaison au FXa. Le rivaroxaban présente à la fois une haute affinité et une bonne biodisponibilité.

Apixaban

Pendant le développement SAR de l’apixaban, trois groupes devaient être testés pour atteindre une puissance et une biodisponibilité maximales. Le premier groupe à tester était le site non actif car il devait être stabilisé avant le test SAR sur le groupe p-méthoxyphényle (fraction de liaison S1). Plusieurs groupes augmentent la puissance du composé, principalement les amides, les amines et les tétrazoles, mais aussi les groupes méthylsulfonyle et trifluorométhyle. Parmi ces groupes, le carboxamide a la plus grande liaison et avait une activité de coagulation similaire à celle des composés.
Dans les tests sur les chiens, ce composé avec un groupe carboxamide appelé 13F, a montré un grand profil pharmacocinétique, une faible clairance et une demi-vie et un volume de distribution adéquats. En raison du succès de la recherche d’un groupe stabilisateur, la recherche SAR pour le groupe de liaison S1 (p-méthoxyphényle) a été abandonnée. Dans le groupe de liaison S4, les analogues N-méthylacétyle et lactame se sont avérés avoir une affinité de liaison très élevée pour le FXa, ont montré une grande coagulation et une grande sélectivité par rapport aux autres protéases. L’orientation s’est avérée importante car le N-méthyl acétyle, comparé à l’acétamide, avait une capacité de liaison au FXa 300 fois plus faible en raison d’une planéité défavorable à proximité du site de liaison de la région S4.
SynthèseEdit
RivaroxabanEdit
Rivaroxaban appartient chimiquement au groupe des n-aryloxazolidinones. Les autres médicaments de ce groupe sont le linézolide et le tedizolide, qui sont tous deux des antibiotiques. Une synthèse de n-aryloxazolidinones à partir d’un éthyl(2,3-dihydroxypropyl)-carbamate protégé par un O-silyle a été publiée en 2016. Dans une réaction monotope, le carbamate se cyclise en un cycle 2-oxazolidone dans des conditions légèrement basiques, tandis que simultanément l’azote de l’oxazolidone est arylisé par catalysation au cuivre. Pour le rivaroxaban en particulier, la 3-morpholinone remplace l’iode en position p du cycle benzénique par une catalyse au cuivre. Ensuite, le groupe protecteur silyle est retiré et l’alcool résultant est remplacé par un groupe amino qui est ensuite acylé dans la dernière étape.
Une préparation industrielle du rivaroxaban a été déposée comme brevet par Bayer Healthcare en 2005. Elle part de la N-(4-aminophénol)-morpholinone qui est alkylée par un dérivé d’oxyde de propylène qui contient également une amine primaire impliquée dans un groupe de protection phtalimide. Ensuite, un équivalent de phosgène est ajouté pour former le cycle 2-oxazolidone et le phtalimide est éliminé. L’amine libre peut maintenant être acylée ce qui conduit au rivaroxaban.
Cependant, selon le brevet, la synthèse présente « divers inconvénients dans la gestion de la réaction qui a des effets particulièrement défavorables pour la préparation ». Le brevet explique également une autre synthèse à partir d’un dérivé du chlorothiophène qui serait plus adaptée au processus industriel mais souligne que des solvants ou des réactifs toxiques doivent être éliminés du produit final. Cette voie n’est donc pas une alternative.
Diverses autres voies de synthèse du rivaroxaban ont été décrites.
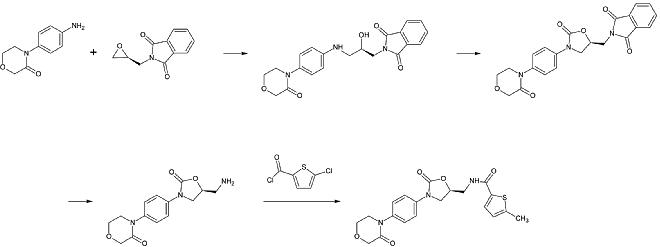
1ère étape : Alkylation de l’amine aromatique primaire
2ème étape : Formation du cycle 2-oxazolinidone, en utilisant un équivalent phosgène
3ème étape : Élimination du groupe de protection phtalimide
4ème étape : Acylation de l’amine primaire
ApixabanEdit
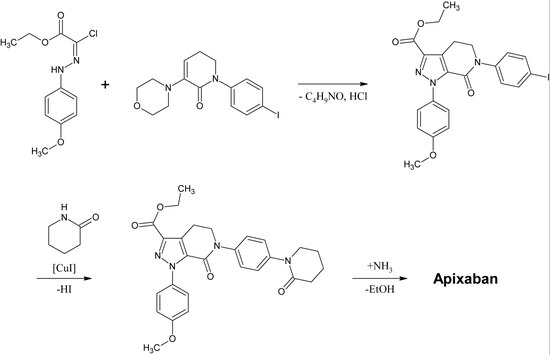
La première synthèse complète de l’apixaban a été publiée en 2007. L’étape clé de cette réaction est une cycloaddition (3+2) d’un dérivé de p-méthoxyphénylchlorohydrazon et d’un dérivé de p-iodophényl-morpholin-dihydropyridine. Après élimination consécutive du HCl et de la morpholine, l’iode est substitué par la 2-pipéridinone par catalyse au cuivre et l’ester éthylique est converti en amide (aminolyse). Cette réaction a été enregistrée comme un brevet en 2009.
.