Descubrimiento y desarrollo de inhibidores directos del Xa
Factor Xa: Estructura y sitios de uniónEditar

Los factores IIa, Xa, VIIa, IXa y XIa son enzimas proteolíticas que tienen un papel específico en la cascada de la coagulación. El factor Xa (FXa) es el más prometedor debido a su posición en la intersección de la vía intrínseca y extrínseca, además de generar alrededor de 1000 moléculas de trombina por cada molécula de Xa, lo que se traduce en un potente efecto anticoagulante. El FXa se genera a partir del FX por escisión de un péptido de activación de 52 aminoácidos, ya que la «a» del factor Xa significa activado. El FXa consta de un dominio catalítico de 254 aminoácidos y también está unido a una cadena ligera de 142 aminoácidos. La cadena contiene el dominio GLA y dos dominios del factor de crecimiento epidérmico (dominios similares al EGF).
El sitio activo del FXa está estructurado para catalizar la escisión de sustratos fisiológicos y escinde PhePheAsnProArg-ThrPhe y TyrIleAspGlyArg-IleVal en la protrombina. El FXa tiene cuatro bolsillos que son objetivos para que los sustratos se unan al factor Xa. Estos bolsillos están alineados por diferentes aminoácidos y los inhibidores del Xa se dirigen a estos bolsillos cuando se unen al factor Xa. Los dos bolsillos más relevantes en cuanto a afinidad y selectividad para los inhibidores del Xa son el S1 y el S4.
S1: El bolsillo S1 es un bolsillo hidrofóbico y contiene un residuo de ácido aspártico (Asp-189) que puede servir como sitio de reconocimiento para un grupo básico. El FXa tiene un espacio residual en el bolsillo S1 y está revestido por los residuos Tyr-228, Asp-189 y Ser-195.
S2: El bolsillo S2 es un bolsillo pequeño y poco profundo. Se fusiona con el bolsillo S4 y tiene espacio para aminoácidos pequeños. La Tyr-99 parece bloquear el acceso a este bolsillo, por lo que este bolsillo no es tan importante como el S1 y el S4.
S3: El bolsillo S3 está situado en el borde del bolsillo S1 y es plano y expuesto al disolvente. Este bolsillo no es tan importante como S1 y S4.
S4: El bolsillo S4 es de naturaleza hidrofóbica y el suelo del bolsillo está formado por el residuo Trp-215. Los residuos Phe-174 y Tyr-99 del FXa se unen a Trp-215 para formar una caja aromática que es capaz de unir fragmentos alifáticos, aromáticos y cargados positivamente. Debido a la unión a entidades cargadas positivamente, puede describirse como un agujero catiónico.
Estructura química y propiedades de los inhibidores directos del XaEditar
| Rivaroxaban | Apixaban | Edoxaban | |
|---|---|---|---|
| MW (g/mol) | 436 | 460 | 548 |
| Fórmula molecular fórmula | C19H18ClN3O5S | C25H25N5O4 | C24H30ClN7O4S |
| Forma | L | L | L |
| Ki | 0.4 nM | 0,08 nM | 0,561 nM |
| IC50 | 0.7 nM | N/A | N/A |
| Biodisponibilidad oral (%) | 66-100 (dosis-dependiente) | 50 | 62 |

Unión de los inhibidores del Xa al factor XaEditar
Todos los inhibidores del Xa se unen en la llamada forma de L dentro del sitio activo del factor Xa. Los componentes clave del factor Xa son los sitios de unión S1 y S4. Se observó por primera vez que los compuestos naturales, la antistasina y el TAP, que poseen componentes altamente polares y, por tanto, cargados, se unen a la diana con cierta especificidad. Por ello, se diseñaron nuevos fármacos con grupos cargados positivamente, pero éstos dieron lugar a una escasa biodisponibilidad. Por lo tanto, los inhibidores de Xa que se comercializan hoy en día contienen un anillo aromático con varios elementos unidos para lograr diferentes interacciones con los sitios de unión S1 y S4. Esto también garantiza una buena biodisponibilidad, además de mantener una firme fuerza de unión. Por lo tanto, los inhibidores del Xa que se comercializan actualmente se basan en enlaces hidrofóbicos y de hidrógeno en lugar de en interacciones altamente polares.
La antistasina se une al factor XaEditar
La antistasina contiene un dominio N- y un dominio C-terminal que son similares en sus secuencias de aminoácidos con un ~40% de identidad y un ~56% de homología. Cada uno de ellos contiene una estructura de hoja β corta y 5 enlaces disulfuro. Sólo el dominio N-terminal es necesario para inhibir el Xa mientras que el dominio C-terminal no contribuye a las propiedades inhibitorias debido a las diferencias en la estructura tridimensional, aunque el dominio C-terminal tiene un patrón fuertemente análogo al sitio activo real.
La interacción de la antistasina con el FXa implica tanto el sitio activo como la superficie inactiva del FXa. El sitio reactivo de la antistasina formado por Arg-34 y Val-35 en el dominio N-terminal se adapta al sitio de unión del FXa, muy probablemente el bolsillo S1. Al mismo tiempo, Glu-15, situado fuera del sitio reactivo de la antistasina, se ajusta a los residuos cargados positivamente en la superficie del FXa. La unión múltiple es termodinámicamente ventajosa y conduce a una inhibición subnanomolar (Ki = 0,3-0,6 nM).
DX-9065a se une al factor XaEditar
DX-9065a, la primera molécula pequeña inhibidora directa del Xa, es un derivado amidinoarílico con un peso molecular de 571,07g/mol. Su grupo amidinonaftaleno cargado positivamente forma un puente salino con el residuo Asp-189 en el bolsillo S1 del FXa. El anillo de pirrolidina encaja entre Tyr-99, Phe-174 y Trp-215 en el bolsillo S4 del FXa.
A diferencia de otros fármacos más antiguos, como la heparina, el DX-9065a es selectivo para el FXa en comparación con la trombina, aunque el FXa y la trombina tengan una estructura similar. Esto se debe a una diferencia en el residuo de aminoácido en la posición 192 del homólogo. Mientras que el FXa tiene un residuo de glutamina en esa posición, la trombina tiene un ácido glutámico que provoca una repulsión electrostática con el grupo carboxilo del DX-9065a. Además, un puente salino entre el Glu-97 de la trombina y el grupo amidina fijado en el anillo de pirrolidina del DX-9065a reduce la flexibilidad de la molécula de DX-9065a, que ahora no puede girar lo suficiente para evitar el choque electrostático. Por ello, el valor IC50 para la trombina es >1000µM mientras que el valor IC50 para el FXa es de 0,16µM.

La unión de rivaroxabán al factor XaEditar
La unión de rivaroxabán al FXa está mediada por dos enlaces de hidrógeno al aminoácido Gly-219. Estos dos enlaces de hidrógeno desempeñan un papel importante al dirigir el fármaco hacia los subsitios S1 y S4 del FXa. El primer enlace de hidrógeno es una interacción fuerte que proviene del oxígeno carbonilo del núcleo de oxazolidinona del rivaroxaban. El segundo enlace de hidrógeno es una interacción más débil y procede del grupo amino de la fracción de carboxamida del clorotiofeno.
Estos dos enlaces de hidrógeno dan lugar a que el fármaco forme una forma de L y encaje en los bolsillos S1 y S4. Los residuos de aminoácidos Phe-174, Tyr-99 y Trp-215 forman un estrecho canal hidrofóbico que constituye el bolsillo de unión S4. La parte de morfolinona del rivaroxabán está «emparedada» entre los aminoácidos Tyr-99 y Phe-174 y el anillo de arilo del rivaroxabán está orientado perpendicularmente a través del Trp-215. El grupo carbonilo de la morfolinona no tiene una interacción directa con la espina dorsal del FXa, sino que contribuye a una planarización del anillo de morfolinona y, por lo tanto, favorece que rivaroxabán se sitúe en sándwich entre los dos aminoácidos.
La interacción entre el sustituyente de cloro de la fracción de tiofeno y el anillo aromático de Tyr-228, que se encuentra en la parte inferior del S1, es muy importante debido a que obvia la necesidad de grupos fuertemente básicos para una alta afinidad por el FXa. Esto permite que el rivaroxabán, que no es básico, alcance una buena biodisponibilidad y potencia oral.

Unión de apixaban al factor XaEditar
Apixaban muestra un modo de unión similar al de rivaroxaban y forma un complejo inhibidor-enzimático estrecho cuando se conecta al FXa. El grupo p-metoxi de apixaban se conecta al bolsillo S1 del FXa pero no parece tener ninguna interacción con ningún residuo de esta región del FXa. El átomo de nitrógeno N-2 del apixaban interactúa con Gln-192 y el oxígeno del carbonilo con Gly-216. El grupo fenil-lactámico de apixaban se sitúa entre Tyr-99 y Phe-174 y, debido a su orientación, es capaz de interactuar con Trp-215 del bolsillo S4. El grupo de oxígeno carbonilo de la fracción de lactama interactúa con una molécula de agua y no parece interactuar con ningún residuo del bolsillo S4.

Relación estructura-actividad (SAR)Edit
Una parte importante del diseño de un compuesto, que es un inhibidor ideal para una determinada diana, es entender la secuencia de aminoácidos del sitio diana para que el compuesto se una. La modelización tanto de la protrombina como del FXa permite deducir la diferencia e identificar los aminoácidos de cada sitio de unión. En la parte inferior del bolsillo S1 del FXa, el aminoácido de unión es Asp-189, al que pueden unirse las amidinas. Después de radiografiar el sitio de unión del FXa, se reveló que el bolsillo S1 tenía una forma plana, lo que significa que un grupo amidinoarilo plano debería unirse a él sin obstáculos estéricos.
Los modernos inhibidores directos del Xa son moléculas en forma de L cuyos extremos encajan perfectamente en los bolsillos S1 y S4. El lado largo de la forma de L tiene que ajustarse a un túnel muy específico dentro del sitio activo de la diana. Para lograrlo, esta parte de las moléculas está diseñada para tener pocas interacciones formales con el FXa en esa región. Como no hay ninguna unión específica, el ajuste de estos agentes entre los bolsillos del FXa aumenta la especificidad total de los fármacos con la molécula del FXa. La interacción entre el bolsillo S1 del FXa y el inhibidor puede ser tanto iónica como no iónica, lo cual es importante porque permite ajustar el diseño de la molécula para aumentar la biodisponibilidad oral. Los compuestos diseñados anteriormente eran moléculas cargadas que no se absorbían bien en el tracto gastrointestinal y, por tanto, no alcanzaban concentraciones séricas elevadas. Los nuevos fármacos tienen una mejor biodisponibilidad, ya que no están cargados y tienen una interacción no iónica con el bolsillo S1.
Rivaroxaban
Durante el desarrollo del SAR del rivaroxaban, los investigadores se dieron cuenta de que añadiendo un grupo de 5-clorotiofeno-2-carboxamida al núcleo de oxazolidonina se podía multiplicar por 200 la potencia, que antes era demasiado débil para su uso médico. Además de este descubrimiento, se confirmó una clara preferencia por la configuración (S). Este compuesto tenía un perfil farmacocinético prometedor y no contenía un grupo amidina altamente básico, pero que anteriormente se había considerado importante para la interacción con el bolsillo S1. Estos hallazgos condujeron a extensas investigaciones de SAR (relación estructura-actividad). Durante las pruebas de SAR, R1 se definió como el grupo más importante para la potencia. La pirrolidinona fue el primer grupo funcional R1 que aumentó significativamente la potencia, pero investigaciones posteriores revelaron una potencia aún mayor con un grupo de morfolinona en su lugar. Los grupos R2 y R3 tenían hidrógeno o flúor y rápidamente se comprobó que el hidrógeno daba lugar a la mayor potencia. A continuación se sustituyeron los grupos R2 y R3 por varios grupos, todos ellos menos potentes que el hidrógeno, por lo que el resultado final fue el hidrógeno. Como la fracción de clorotiofeno tenía una solubilidad inadecuada en el agua, se intentó sustituirla por otro grupo, pero no tuvo éxito. La fracción de clorotiofeno se une a la Tyr-228 en la parte inferior del bolsillo S1, lo que la convierte en un factor clave para la unión al FXa. Rivaroxaban tiene tanto una alta afinidad como una buena biodisponibilidad.

Apixaban

Durante el desarrollo del SAR de apixaban había tres grupos que debían probarse para obtener la máxima potencia y biodisponibilidad. El primer grupo que se probó fue el sitio no activo, ya que es necesario estabilizarlo antes de las pruebas de SAR en el grupo p-metoxifenilo (fracción de unión S1). Hay varios grupos que aumentan la potencia del compuesto, principalmente amidas, aminas y tetrazoles, pero también grupos metilsulfonilo y trifluorometilo. De estos grupos, la carboxamida es el que más se une y tuvo una actividad coagulante similar a la de los compuestos.
En las pruebas con perros, este compuesto con un grupo carboxamida llamado 13F, mostró un gran perfil farmacocinético, un bajo aclaramiento y una adecuada vida media y volumen de distribución. Debido al éxito de encontrar un grupo estabilizador, se suspendió la investigación del SAR para el grupo de unión S1 (p-metoxifenilo). En el grupo de unión S4, los análogos N-metilacetilo y lactámicos demostraron tener una afinidad de unión muy alta por el FXa, mostrando una gran coagulación y selectividad frente a otras proteasas. La orientación resultó ser importante, ya que el N-metilacetilo, en comparación con la acetamida, tenía una capacidad de unión al FXa 300 veces menor debido a una planaridad desfavorable cerca del sitio de unión de la región S4.
SíntesisEditar
RivaroxabanEditar
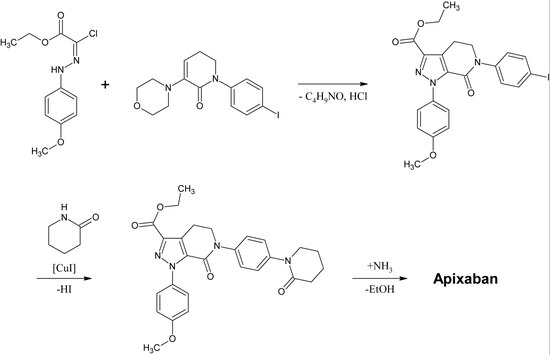
Rivaroxaban pertenece químicamente al grupo de las n-aryloxazolidinonas. Otros fármacos de ese grupo son el linezolid y el tedizolid, ambos antibióticos. En 2016 se publicó una síntesis de n-ariloxolidinonas a partir de un etil(2,3-dihidroxipropil)-carbamato protegido con O-sililo. En una reacción de una sola vez, el carbamato se cicliza a un anillo de 2-oxazolidona en condiciones ligeramente básicas, mientras que simultáneamente el nitrógeno de la oxazolidona se arila mediante la catalización con cobre. Para el rivaroxaban en particular, la 3-morfolinona sustituye el yodo en la posición p del anillo de benceno mediante la catalización con cobre. Después, se elimina el grupo protector sililo y el alcohol resultante se sustituye por un grupo amino que se atila en el último paso.
Una preparación industrial de rivaroxaban fue registrada como patente por Bayer Healthcare en 2005. Parte de la N-(4-aminofenol)-morfolinona que es alquilada por un derivado de óxido de propileno que también contiene una amina primaria implicada en un grupo de protección de ftalimida. A continuación, se añade un equivalente de fosgeno para formar el anillo de 2-oxazolidona y se elimina la ftalimida. La amina libre puede ahora ser acilada, lo que conduce al rivaroxaban.
Sin embargo, según la patente la síntesis tiene «varias desventajas en la gestión de la reacción que tiene efectos particularmente desfavorables para la preparación». La patente también explica otra síntesis a partir de un derivado de clorotiofeno que sería más adecuada para el proceso industrial, pero señala que hay que eliminar disolventes o reactivos tóxicos del producto final. Por lo tanto, esta vía no es una alternativa.
Se han descrito varias otras vías de síntesis de rivaroxaban.
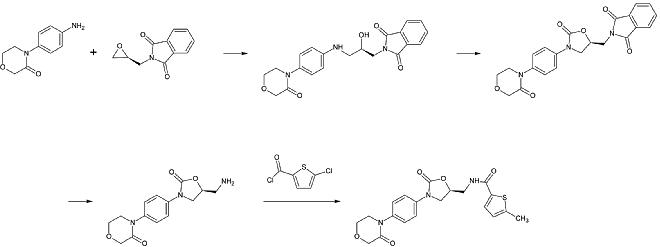
1er paso: Alquilación de la amina aromática primaria
Segundo paso: Formación del anillo de 2-oxazolinidona, utilizando un equivalente de fosgeno
3ª etapa: Eliminación del grupo protector de la ftalimida
4ª etapa: Acilación de la amina primaria
ApixabanEditar
La primera síntesis completa de apixaban se publicó en 2007. El paso clave de esta reacción es una (3+2)cicloadición de un derivado de p-metoxifenilclorhidrazón y un derivado de p-yodofenil-morfolina-dihidropiridina. Tras la siguiente eliminación del HCl y la morfolina, el yodo se sustituye por la 2-piperidinona mediante una catalización de cobre y el éter etílico se convierte en una amida (aminólisis). Esta reacción se registró como patente en 2009.