A fehérállomány-rendellenességek diagnosztikai képalkotása | Veleszületett agyi hipomielináció; Pelizaeus-Merzbacher-kór és a kapcsolódó rendellenességek hálózata
Diagnosztic Imaging of White Matter Abnormalities
Junichi TAKANASHI, Gyermekgyógyászati Tanszék, Tokyo Women’s Medical University, Yachiyo Medical Center
Bevezetés
Ez a cikk a mágneses rezonancia képalkotáson (MRI) az agyi fehérállományban rendellenes jelek formájában megjelenő rendellenességek diagnosztizálására alkalmazott megközelítést vázolja fel a képalkotástól a diagnózisig. A főként a fehérállományt érintő rendellenességeket angolul általában “leukoencephalopathia” vagy “white matter disorders” néven emlegetik1, 2) 3) Egy másik kifejezést, a leukodisztrófiát néha összekeverik a fehérállomány-degenerációval, de ez valójában a genetikai komponenssel rendelkező rendellenességek (örökletes demyelinizáló rendellenességek) szűkebb spektrumára utal.
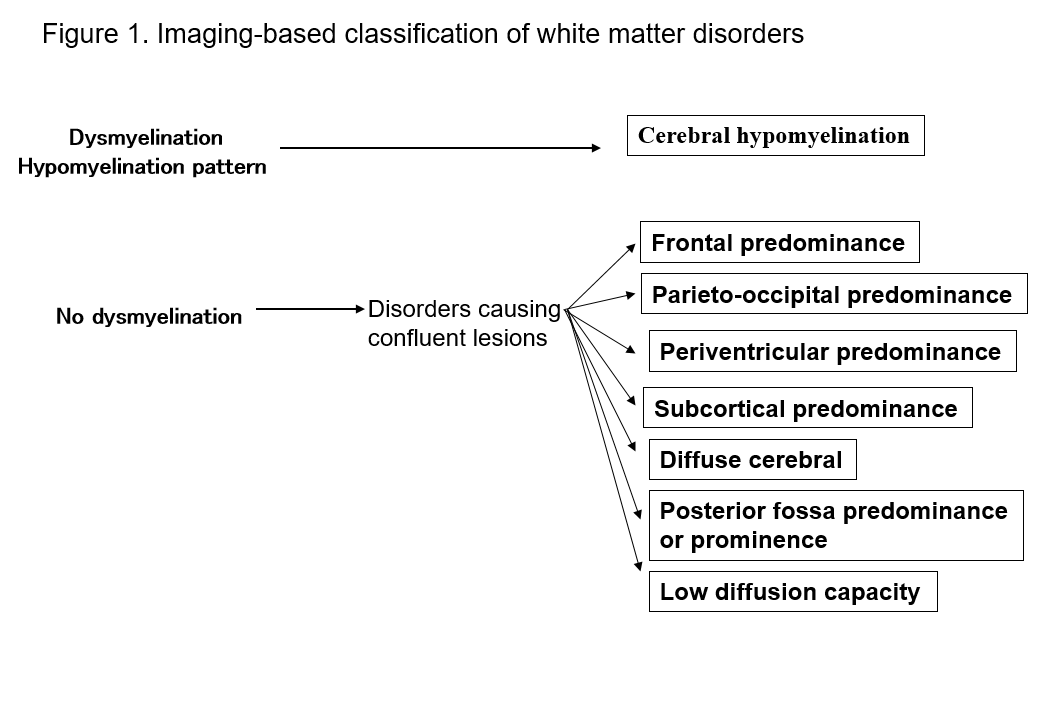
A fehérállomány-rendellenességek képalkotáson alapuló osztályozása
Az MRI megjelenése drámaian javította a központi idegrendszer fehérállományában lévő elváltozások kimutatására való képességünket. A fehérállomány-rendellenességek számos ismert formája specifikus jeleket mutat az MRI-n, ami hasznos a diagnózisukhoz. Az MRI-n (T1-súlyozott, T2-súlyozott vagy FLAIR-képalkotás) látható fehérállomány-rendellenességek mintázatainak azonosítása megkönnyíti a lehetőségek leszűkítését a többféle differenciáldiagnózisban. Schiffmann és van der Kamp fehérállomány-rendellenességek MRI-lelet szerinti osztályozása gyakorlati értékkel bír2, 4) (1. ábra, 1. táblázat). Még ha ez nem is vezet végleges diagnózishoz, a képalkotó leletek osztályozása később egy új rendellenesség felfedezéséhez vezethet. Az alábbiakban a fehérállományi rendellenességeket a fenti MRI-osztályozások alapján ismertetem, és a rendellenességek főbb típusait magyarázom.
1. táblázat. Rendellenességek listája MRI-minták alapján
- Frontális predominancia
Alexander-kór, az X-kapcsolt adrenoleukodisztrófia (ALD) frontális változata, metakromatikus leukodisztrófia (MLD), neuroaxonális leukodisztrófia szferoidokkal. - Parieto-occipitalis predominancia
X-hez kötött adrenoleukodisztrófia (ALD), Krabbe-kór,korai kezdetű peroxiszomális rendellenességek, újszülöttkori hipoglikémia. - Periventrikuláris predominancia
Metakromatikus leukodisztrófia (MLD), Krabbe-kór, Sjögren-Larsson-szindróma, felnőttkori poliglükozán test betegség, leukoencephalopathia agytörzsi és gerincvelői érintettséggel és laktátemelkedéssel (LBSL), periventrikuláris leukomalacia (PVL), HIV encephalopathia, késői kezdetű neuronális ceroid lipofuscinózisok. - Subcorticalis predominancia
L-2-hidroxiglutársavuria, galaktozémia, Kearns-Sayer-szindróma, propionakadémia, karbamidciklus-zavarok, Canavan-kór. - Diffúz agyi
Megalencephalicus leukoencephalopathia subcorticalis cisztákkal (MLC), leukoencephalopathia eltűnő fehér anyaggal (VWM), merozinhiányos veleszületett izomdystrophia, mitokondriális betegség, molibdén kofaktorhiány, szulfitoxidázhiány, fehérállomány-rendellenességek előrehaladott esetei. - A hátsó fossa túlsúlya vagy kiemelkedése
A kisagy és a kisagyi pedunculusok léziói: cerebrotendinosus xanthomatosis (CTX), peroxisomális rendellenességek, Alexander-kór, leukoencephalopathia agytörzsi és gerincvelői érintettséggel és laktátemelkedéssel (LBSL), juharszirupos vizeletbetegség, histiocytosis, felnőttkori autoszomális domináns leukodisztrófia, amely lamin B1 duplikációhoz kapcsolódik, heroin- és kokainmérgezés.
Agytörzsi elváltozások: Alexander-kór, LSBL, peroxiszomális rendellenességek, Wilson-kór, felnőttkori poliglükozán betegség, Leigh-szindróma, dentatorubropallidoluiloszi atrófia (DRPLA), felnőttkori poliglükozán test betegség, lamin B1 duplikációval összefüggő felnőttkori autoszomális domináns leukodisztrófia. - Multifokális elváltozások
TORCH-szindróma (veleszületett citomegalovírus-fertőzés), brucellózis, akut disszeminált encephalomyelitis (ADEM), szklerózis multiplex (MS), neuromyelitis optica (NMO), agyi autoszomális domináns arteriopátia szubkortikális infarktusokkal és leukoencephalopathiával (CADASIL), ateroszklerózis, amiloid angiopátia, COL4A1-asszociált agyi kisérbetegség, Fabry-kór, Susac-szindróma, mitokondriális betegség, L-2-hidroxiglutársav-uria, mukopoliszacharidózis (MPS), kromoszóma-rendellenességek (pl. 6p-szindróma). - Alacsony diffúziós kapacitású elváltozások
A juharszirupos vizeletbetegség, metionin adenozil-transzferáz I/III hiány, fenilketonúria, nem ketotikus hyperglicinémia, Canavan-kór, aktív elváltozások Krabbe-kórban és metakromatikus leukodisztrófia.
1. Agyi fehérállomány hipomielinizáció
A rendellenességek azon csoportjára utal, amelyben a myelinhüvely kialakulása károsodott vagy késik, és képei az éretlen myelinizációjú újszülöttekéhez hasonlítanak. A T2-súlyozott képeken a fehérállomány jellemzően kiterjedt hiperintenzitásként jelenik meg, amely a kéreghez képest halvány. További részletekért lásd a veleszületett agyi hipomielináció honlapját.
Ha a fehérállományi elváltozások nem felelnek meg az agyi fehérállomány hipomielinációnak, meg kell határozni, hogy konfluensek vagy multiplexek.2) A konfluens fehérállományi elváltozások általában öröklött fehérállomány-degeneráció (leukodisztrófia) következményei, és a legtöbb esetben kétoldali szimmetrikusak. A multiplex fehérállomány-elváltozások általában aszimmetrikusak és szerzettek. A konfluens fehérállományi elváltozásokat az alábbi 2-7. kategóriákra osztjuk tovább.
2. Frontális predomináció
A rendellenességek ezen csoportjában a kiterjedt fehérállományi elváltozások túlnyomórészt a homloklebenyben vannak jelen. Ide tartozik az Alexander-kór, az X-kapcsolt adrenoleukodisztrófia (ALD) frontális változata, a metakromatikus leukodisztrófia (MLD) és a szferoidokkal járó neuroaxonális leukodisztrófia.
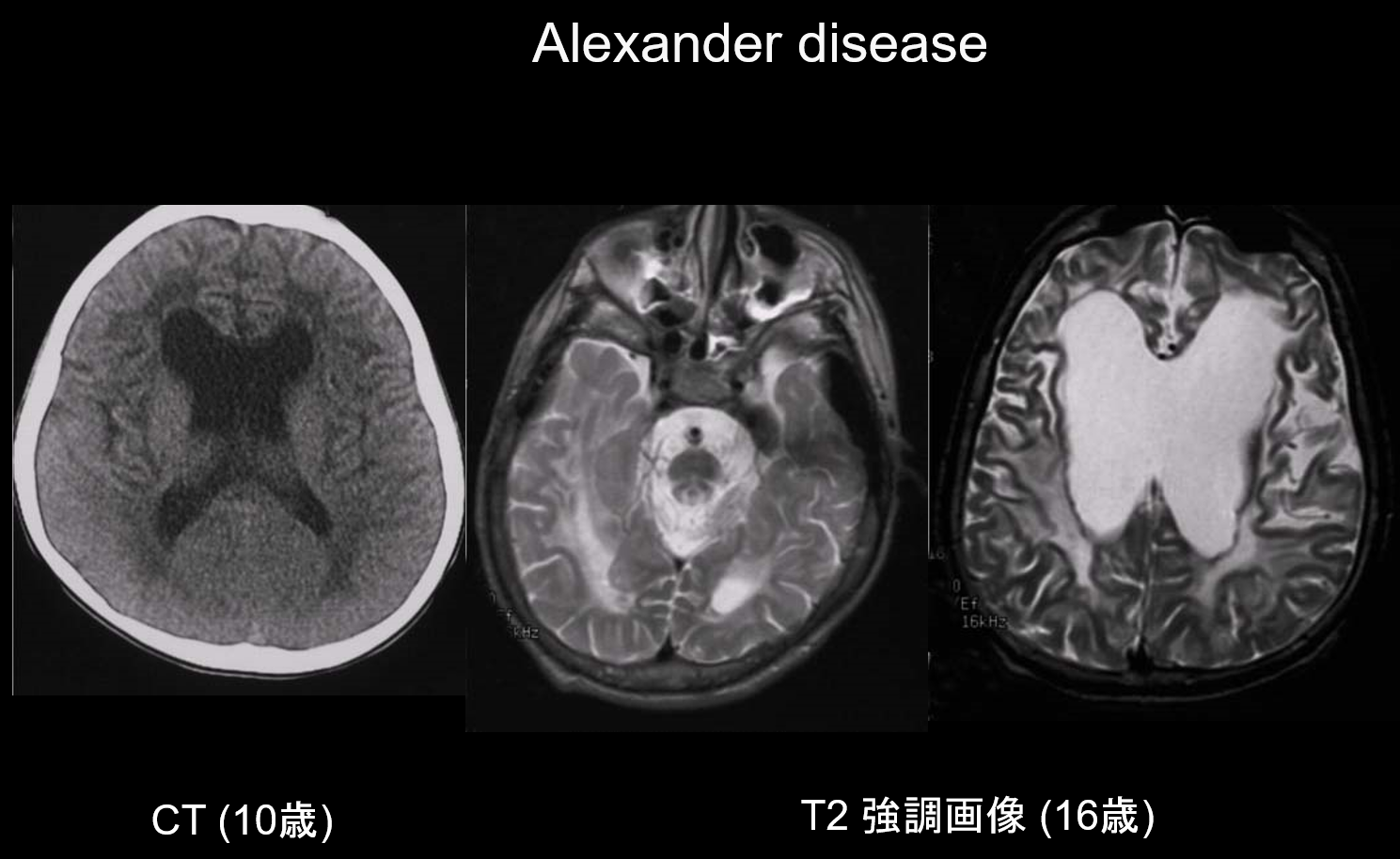
(a) Alexander-kór.
Az Alexander-kór autoszomális dominánsan öröklődő rendellenesség, amelyet a 17q21 kromoszómán található GFAP gén mutációja okoz. A Rosenthal-rostok felhalmozódását eredményezi a sztellás gliasejtekben. Ezek a rostok GFAP-ból és stresszfehérjékből (αB-kristallin és HSP27. Az Alexander-kór főként csecsemőkorban, 3 hónapos és 2 éves kor között jelentkezik, megalenkefália, fejlődési elmaradás, spasztikus bénulás és epilepszia megjelenésével. MRI-n (i) kiterjedt fehérállományi elváltozásokat mutathat, túlnyomórészt a homloklebenyben; (ii) T1 hiperintenzív és T2 hipointenzív marginalizáció az oldalkamrák körül; (iii) elváltozások a bazális ganglionokban és a talamuszban; (iv) agytörzsi elváltozások; és (v) az aktív elváltozások kontrasztos erősödése (2. ábra). A korai stádiumban a fehérállomány és a putamen elváltozásai mellett duzzanat látható, amely fokozatosan atrófiát vagy cisztaképződést okozhat.
3. Parieto-occipitalis predominancia
A rendellenességek e csoportjának fő jellemzője a parieto-occipitalis fehérállomány elváltozás. Ide tartozik az X-hez kötött adrenoleukodisztrófia (ALD), a Krabbe-kór, a korai peroxisomális rendellenességek és az újszülöttkori hipoglikémia.
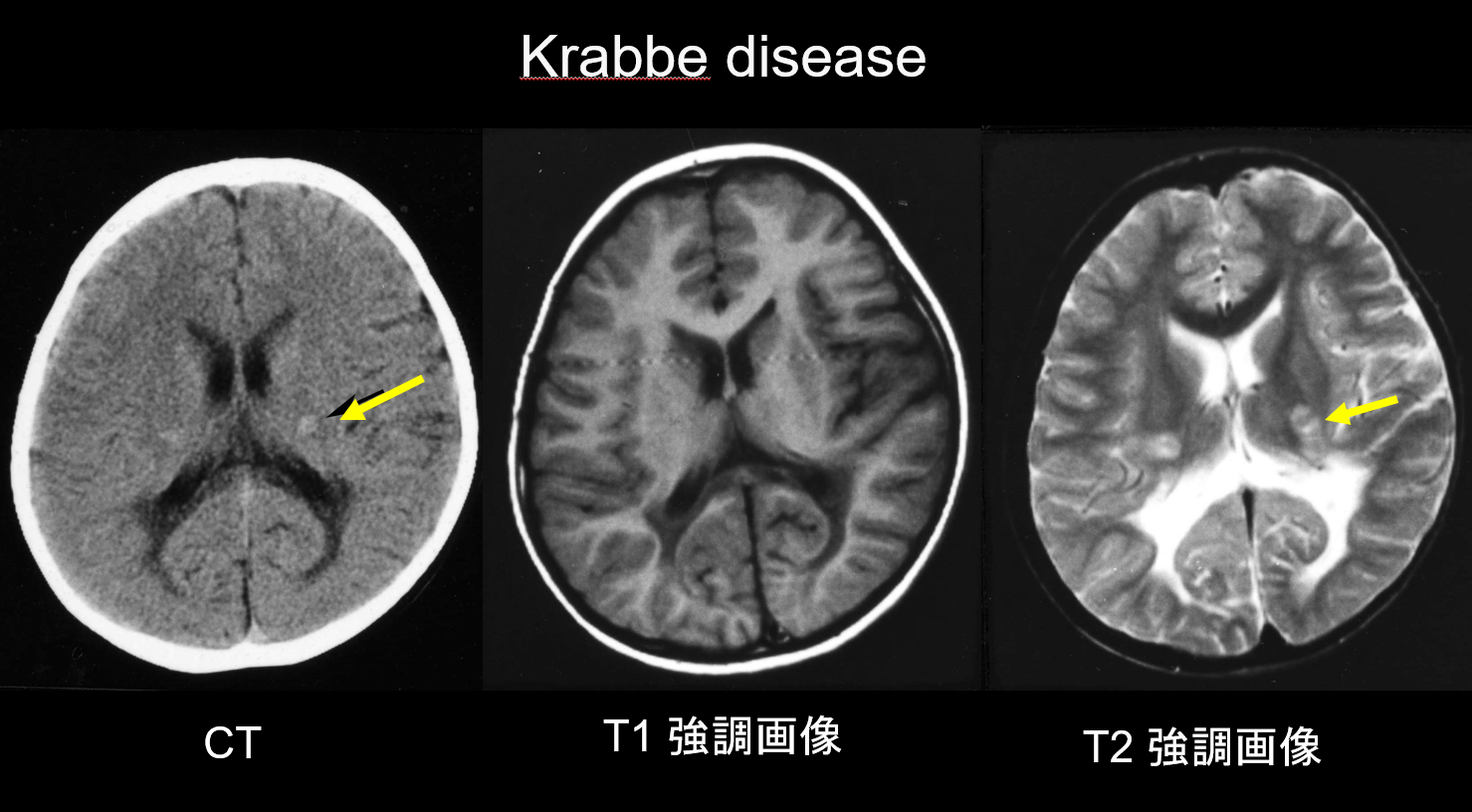
(a) Krabbe-kór.
A Krabbe-kór egy autoszomális recesszív öröklődő rendellenesség (lizoszomális tárolási betegség), amelyet galaktozilceramidáz-hiány okoz (14q31 kromoszóma), és amelyben az erősen citotoxikus pszichozin felhalmozódása feltételezhetően kiterjedt demielinációt okoz. Nagy, többmagvú sejtek, úgynevezett “globoid sejtek” is megjelennek. Attól függően, hogy milyen életkorban jelenik meg, infantilis, késői kezdetű infantilis, juvenilis kezdetű vagy felnőtt kezdetű betegségnek minősül. A legtöbb eset csecsemőkori, és 3-6 hónapos korban láz, ingerlékenység, táplálkozási nehézségek, fejlődési elmaradás, perifériás neuropátia, spaszticitás és látóideg-atrófia megjelenésével kezdődik. A korai stádiumban a komputertomográfia (CT) jellegzetes hiperdenzitást mutat a thalamusban és a corona radiatában. Ez feltehetően nagy sűrűségű globoid sejteket és gliaproliferációt tükröz. Az MRI T1 hiperintenzitást és T2 hipointenzitást is mutathat a kamrák körül, valamint az MLD-ben látottakhoz hasonló lineáris struktúrákat (3. ábra). A kisagyi fogazott mag, a kisagyi fehérállomány és az agytörzsi piramispálya már korai stádiumban T2 hiperintenzitást mutat.
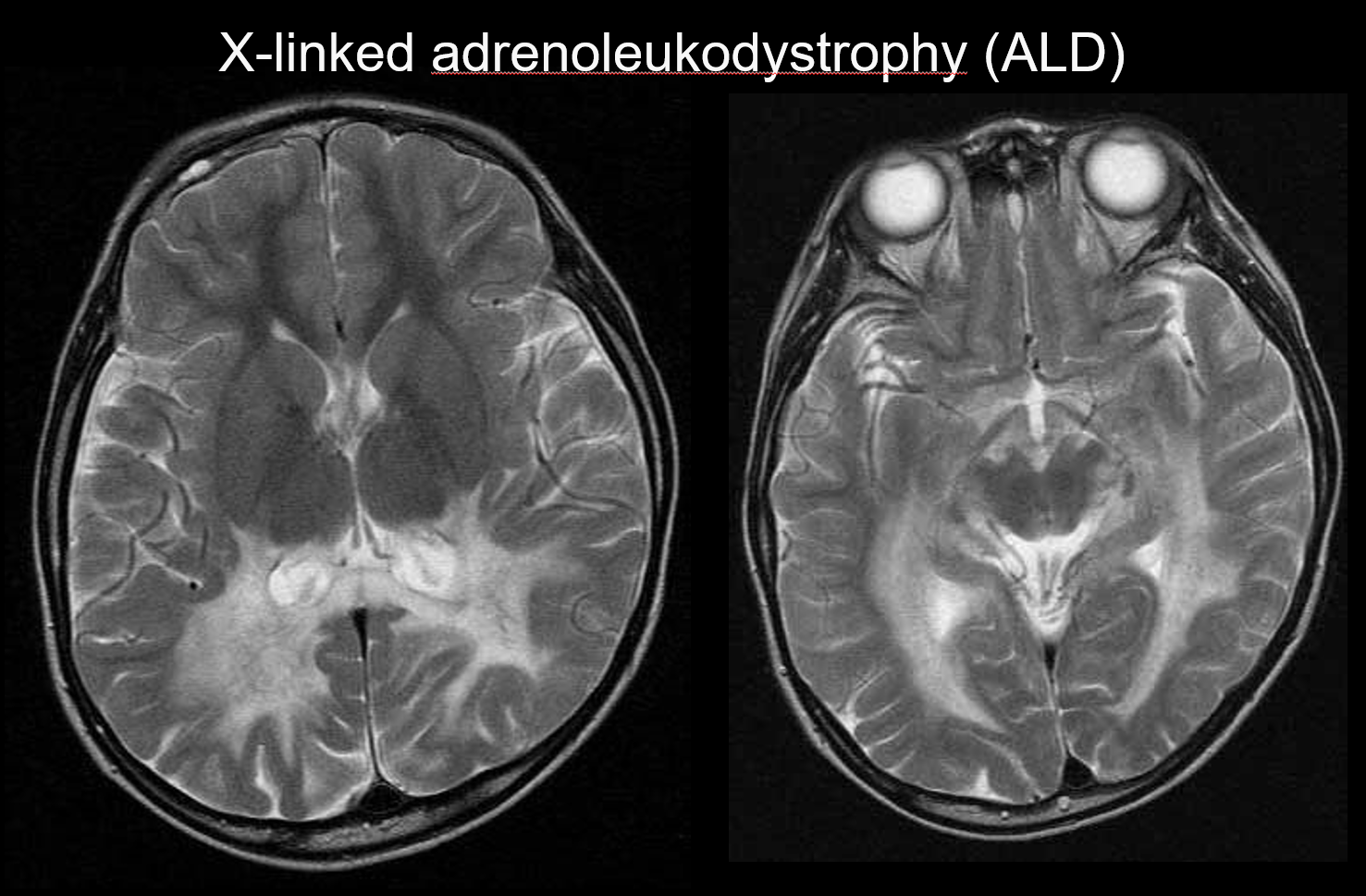
(b) X-hez kötött adrenoleukodisztrófia
A X-hez kötött adrenoleukodisztrófia (ALD) egy X-hez kötött recesszív öröklődő rendellenesség (peroxiszomális rendellenesség), amelyet az ABCD1 gén (Xq28 kromoszóma) rendellenessége okoz. A károsodott β-oxidáció a nagyon hosszú láncú zsírsavak felhalmozódását eredményezi az agyi fehérállományban és a mellékvesékben, ami demyelinizációt és mellékvesekéreg-elégtelenséget okoz. Az ALD-t gyermekkori, serdülőkori és felnőttkori agyi formákra, adrenomyeloneuropátiára (AMN) és kizárólag Addison-kórra osztják. A gyermekkori cerebrális forma 5-8 éves korban alakul ki, a tünetek megjelenésével, beleértve az intellektuális hanyatlást, a spasztikus járást, valamint a látás- és halláskárosodást. Patológiailag a demielinizáció az oldalsó kamra trigonusát körülvevő fehérállománytól a corpus callosum spleniumáig terjed, fokozatosan anterolaterálisan terjeszkedve. A betegség patológiáját tükröző, szimmetrikus T2 hiperintenzitások és T1 hipointenzitások jelennek meg az MRI-n, amelyek anterolaterálisan terjednek az oldalkamra trigonusát körülvevő fehérállományból, és a peremeken kontrasztfokozódás látható (4. ábra). A kortikospinális pálya elváltozásai is nyilvánvalóak.
4. Periventricularis predominancia
Ezekre a rendellenességekre elsősorban az oldalkamrákat körülvevő fehérállomány elváltozásai jellemzőek, a szubkortikális fehérállomány (U-rostok) megmaradnak. Ez a mintázat számos rendellenességben megfigyelhető, beleértve az MLD-t is, és ezért viszonylag nem specifikus. Az oldalkamrák körüli enyhe kóros jelek a kérgi degenerációban is megfigyelhetők, különösen a gyermekkor után kialakuló neuronális ceroid lipofuscinózisokban.
(a) Metakromatikus leukodisztrófia.
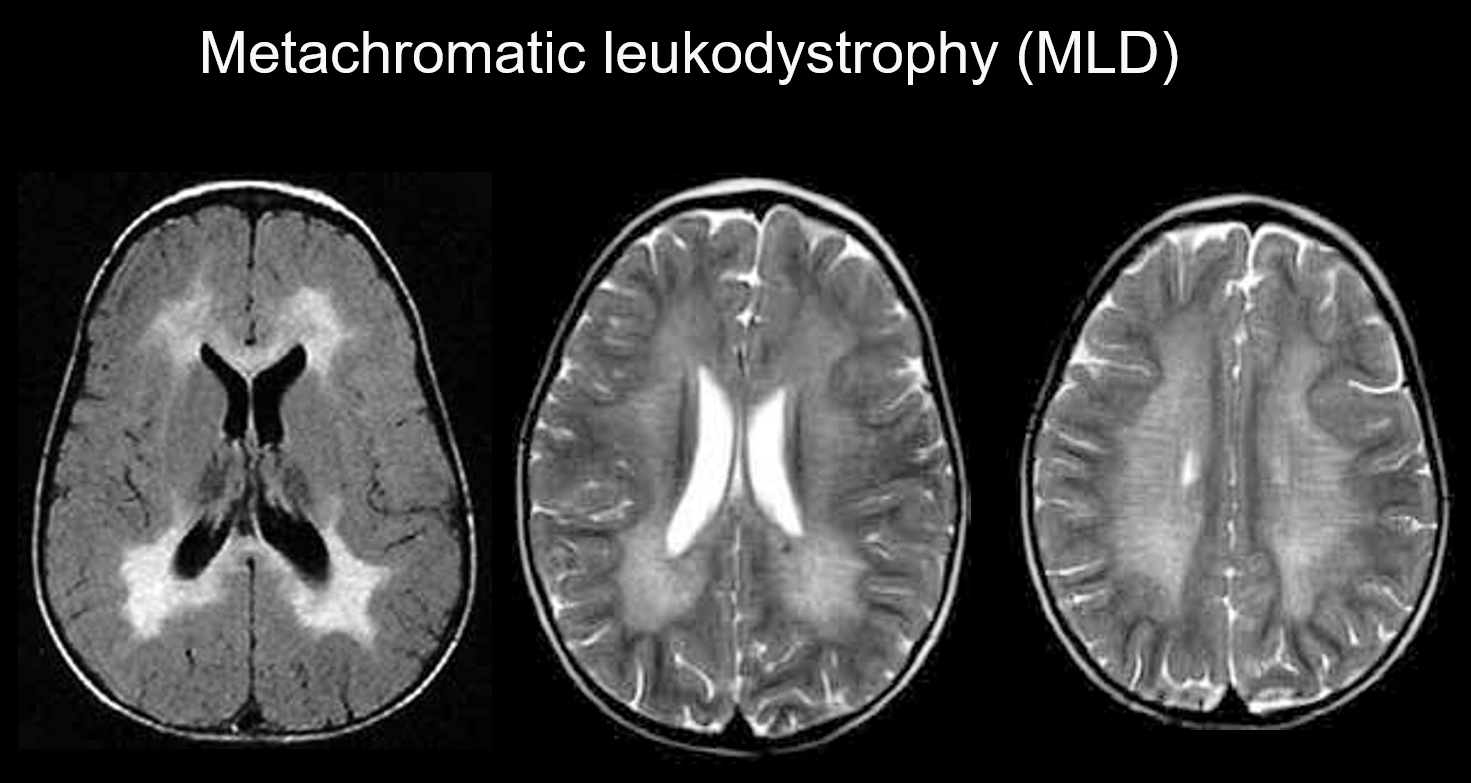
A metakromatikus leukodisztrófia egy autoszomális recesszív öröklődő betegség (lizoszomális tárolási rendellenesség), amelyet az arilszulfatáz-A hiánya okoz (22q13.31 kromoszóma), amelyben az erősen toxikus szulfatid felhalmozódása demielinációt eredményez. Attól függően, hogy milyen életkorban jelentkezik, veleszületett, csecsemőkorban kialakuló, fiatalkorban kialakuló vagy felnőttkorban kialakuló betegségnek minősül. Tünetei közé tartozik a kognitív regresszió, a spasztikus bénulás, az önkéntelen mozgások, a perifériás neuropátia és a látóideg sorvadása. T2-súlyozott képalkotáson a fehérállomány hiperintenzitásaként jelenik meg, főként az oldalsó kamrák körül, T1-súlyozott képalkotáson pedig enyhe hipointenzitásként. Az elváltozások túlnyomórészt a homloklebenyben fordulnak elő. A fehérállományban a kiterjedt kóros jeleken belül normális intenzitású sávok (tigriscsíkok) is megjelenhetnek (5. ábra). Ezek feltehetően a myelinhüvely részleges megőrzésének köszönhetőek a perivascularis térben és a myelinhüvely bomlástermékeinek felhalmozódásának a makrofágokban.
5. Subkortikális túlsúly
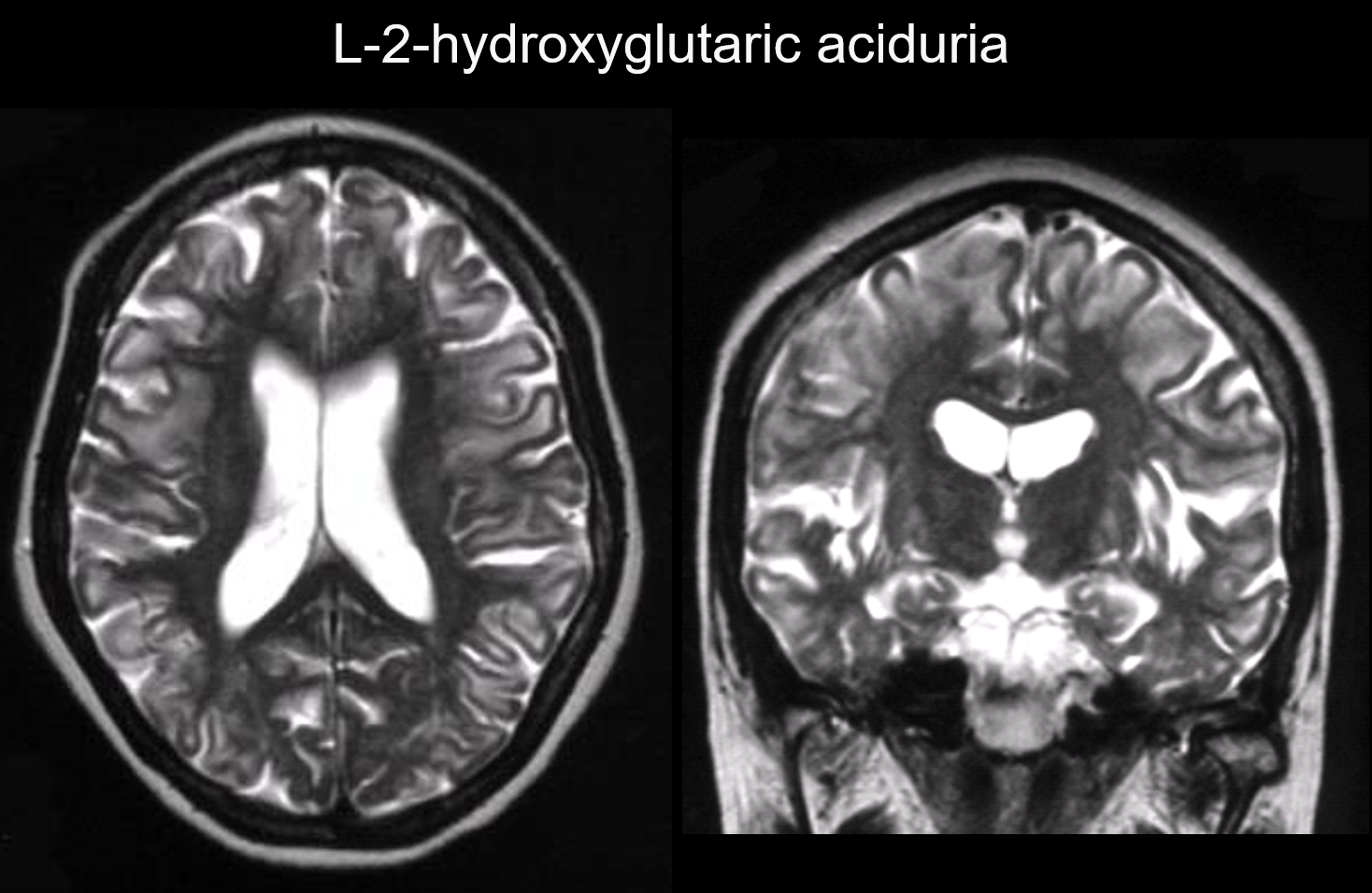
Ezekben a rendellenességekben az elváltozások elsősorban a szubkortikális fehérállományban fordulnak elő, beleértve az U-rostokat is. Az ilyen mintázatú rendellenességek közé tartozik az L-2-hidroxiglutársavuria (6. ábra), a galaktozémia, a Kearns-Sayer-szindróma, a propionakadémia, a karbamidciklus zavarai és a korai stádiumú Canavan-kór.
6. Diffúz agyi
Ezekben a rendellenességekben az egész agyi fehérállományban kóros jelek jelennek meg. Ezek erős T2 hiperintenzitást mutatnak a nem myelinizált fehérállomány által keltett T2 jelekhez képest (hypomyelinizáció). A szubkortikális cisztákkal járó megalencephalicus leukoencephalopathia és az eltűnő fehérállományú leukoencephalopathia esetein kívül a fehérállomány bármilyen típusú rendellenességében szenvedő betegek a betegség előrehaladtával végül ezt a mintázatot mutatják.
(a) Megalencephalicus leukoencephalopathia subcorticalis cisztákkal (MLC)
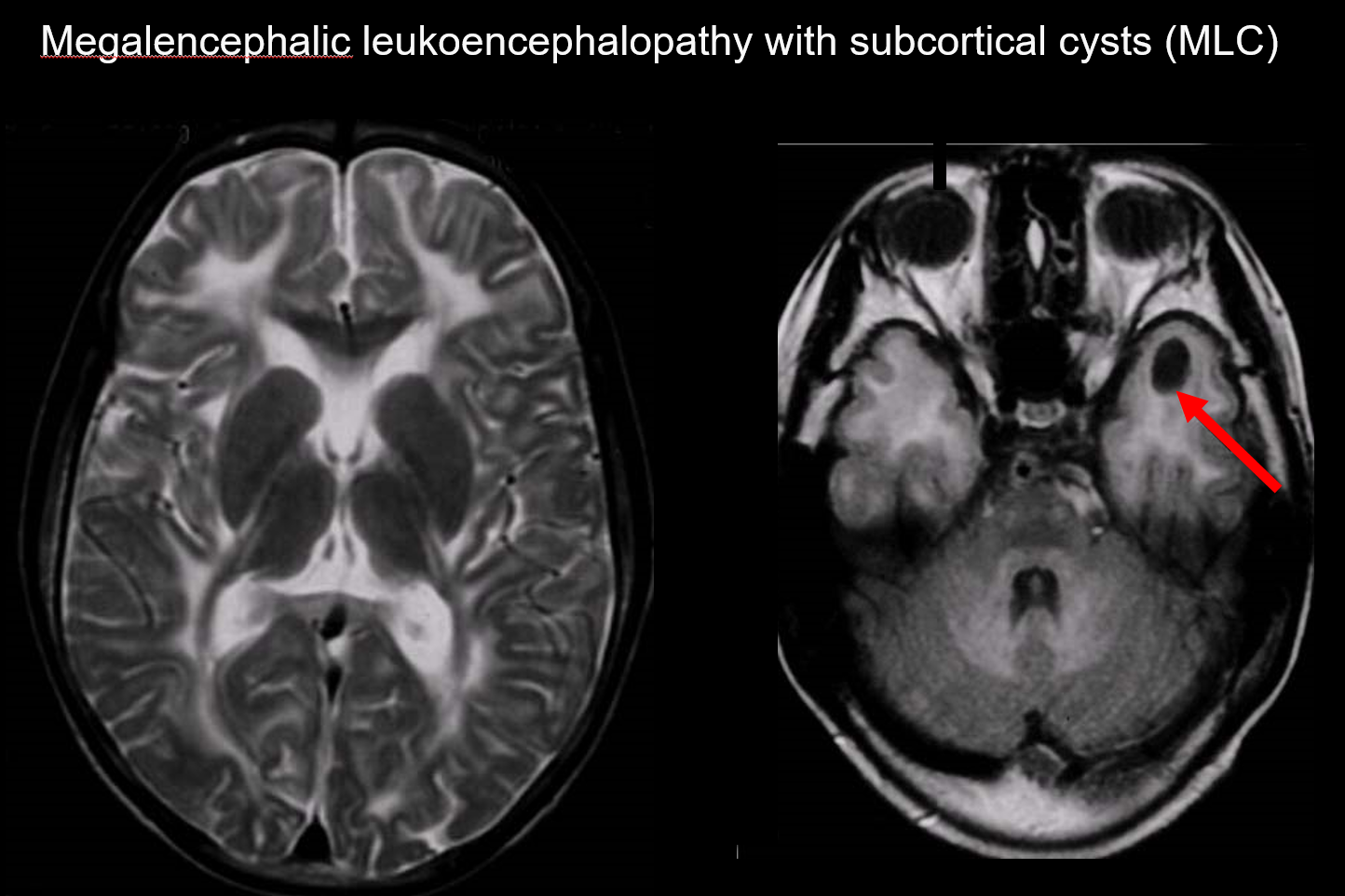
A MLC az MLC1 gén rendellenessége által okozott autoszomális recesszív öröklődő rendellenesség, amelynek csecsemőkori kezdetét megalocephalia, lassan előrehaladó motoros leépülés, ataxia és spaszticitás jellemzi. Az MRI jellegzetes, széles körben elterjedt kóros jeleket mutat a fehérállományban és a fehérállomány enyhe duzzanatát, valamint cisztaképződést a parietális és temporális lebenyekben (7., 8. ábra).7, 8) A T1-súlyozott és T2-súlyozott képalkotás kóros fehérállományt mutat, míg a ciszták mind T1 hipointenzitást és T2 hiperintenzitást mutatnak, ami különösen megnehezíti a kimutatásukat. A FLAIR képalkotás, amely a cisztákat (vizet) hipointenzitásként ábrázolja, értékes a diagnózisához. Japánok körében gyakoribb, mint a vacuoláló megalencephalicus leukoencephalopathia.
(b) Leukoencephalopathia eltűnő fehér anyaggal.
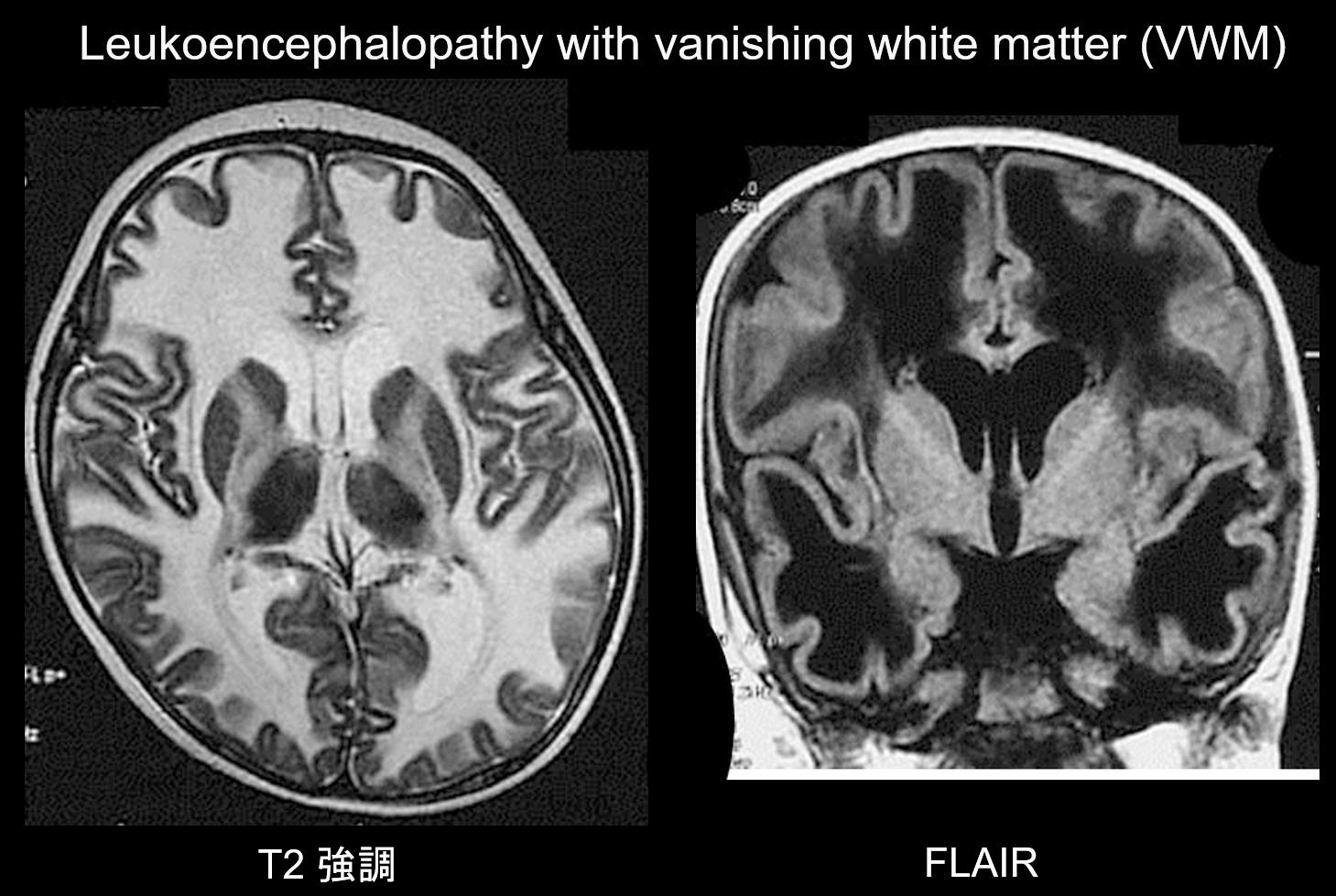
Az eltűnő fehér anyaggal járó leukoencephalopathia (VWM) egy autoszomális recesszív öröklődő rendellenesség, amelyet az eIF2B, az eIF2-hez kapcsolódó fehérje hiánya okoz, amely az iniciátor tRNS-t a riboszómákra továbbítja. Az eIF2B öt különböző fehérjéből áll, amelyek mind különböző genetikai lókuszokkal rendelkeznek. Kimutatták, hogy a VWM ugyanaz a rendellenesség, mint a gyermekkori kisagyi ataxia és a centrális hipomielinizáció (CACH). A betegek az újszülöttkorban és a korai csecsemőkorban normálisak, de a betegség kezdete után (általában 2-6 éves korban) lassan progresszív kognitív regresszió, spaszticitás és ataxia alakul ki náluk. Ezek a tünetek köztudottan súlyosbodnak fertőzés vagy kisebb trauma hatására. Az agyi fehérállomány kiterjedt T2 hiperintenzitást és T1 hipointenzitást mutat, és idővel fokozatosan folyadékkal helyettesíti (ahogy a név is jelzi, a fehérállomány eltűnik) (8. ábra). A cisztás fehérállomány sávos struktúrákat tartalmaz, amelyek feltehetően a megmaradt szövetet képviselik. Az agytörzsben, különösen a központi tegmentális traktusban is észlelhetők kóros jelek. A FLAIR képalkotás értékes e rendellenesség diagnosztizálásában.
7. A hátsó fossa túlsúlya vagy kiemelkedése
Ezekre a rendellenességekre jellemzőek a túlnyomórészt az agytörzsben és a kisagyban található elváltozások. A kisagy fehérállományának elváltozásait olyan rendellenességek okozhatják, mint a cerebrotendinosus xanthomatosis (CTX), peroxisomális rendellenességek, Alexander-kór, leukoencephalopathia agytörzsi és gerincvelői érintettséggel és laktátemelkedéssel (LBSL), juharszirupos vizeletbetegség, histiocytosis, valamint heroin- és kokainmérgezés. Az agytörzsi elváltozásokat olyan rendellenességek okozhatják, mint az Alexander-kór, az LSBL és a felnőttkori poliglükóz betegség. A középső kisagyi törzs elváltozásai a törékeny X-szindrómában és a lamin B1 duplikációval összefüggő felnőtt autoszomális domináns leukodisztrófiában fordulnak elő.
8. Multifokális elváltozások
A fenti 2-7. pontban leírt konfluens elváltozásokat okozó rendellenességektől eltérően az ebben a szakaszban szereplő rendellenességek multifokális (szétszórt) elváltozásokat eredményeznek. Ide tartoznak az olyan fertőzések, mint a TORCH-szindróma (veleszületett citomegalovírus-fertőzés vagy más ok miatt) és a brucellózis; gyulladásos betegségek, mint az akut disszeminált encephalomyelitis (ADEM), a sclerosis multiplex (MS) és a neuromyelitis optica (NMO); vaszkulopátiák, mint az agyi autoszomális domináns arteriopátia szubkortikális infarktusokkal és leukoencefalopátiával (CADASIL), ateroszklerózis, amiloid angiopátia, COL4A1-asszociált agyi kisérbetegség, Fabry-kór és Susac-szindróma; és olyan öröklött állapotok, mint a mitokondriális betegség, az L-2-hidroxiglutársav-uria, a mukopoliszacharidózis (MPS) és a kromoszóma-rendellenességek (például a 6p-szindróma).
9. Alacsony diffúziós kapacitású elváltozások
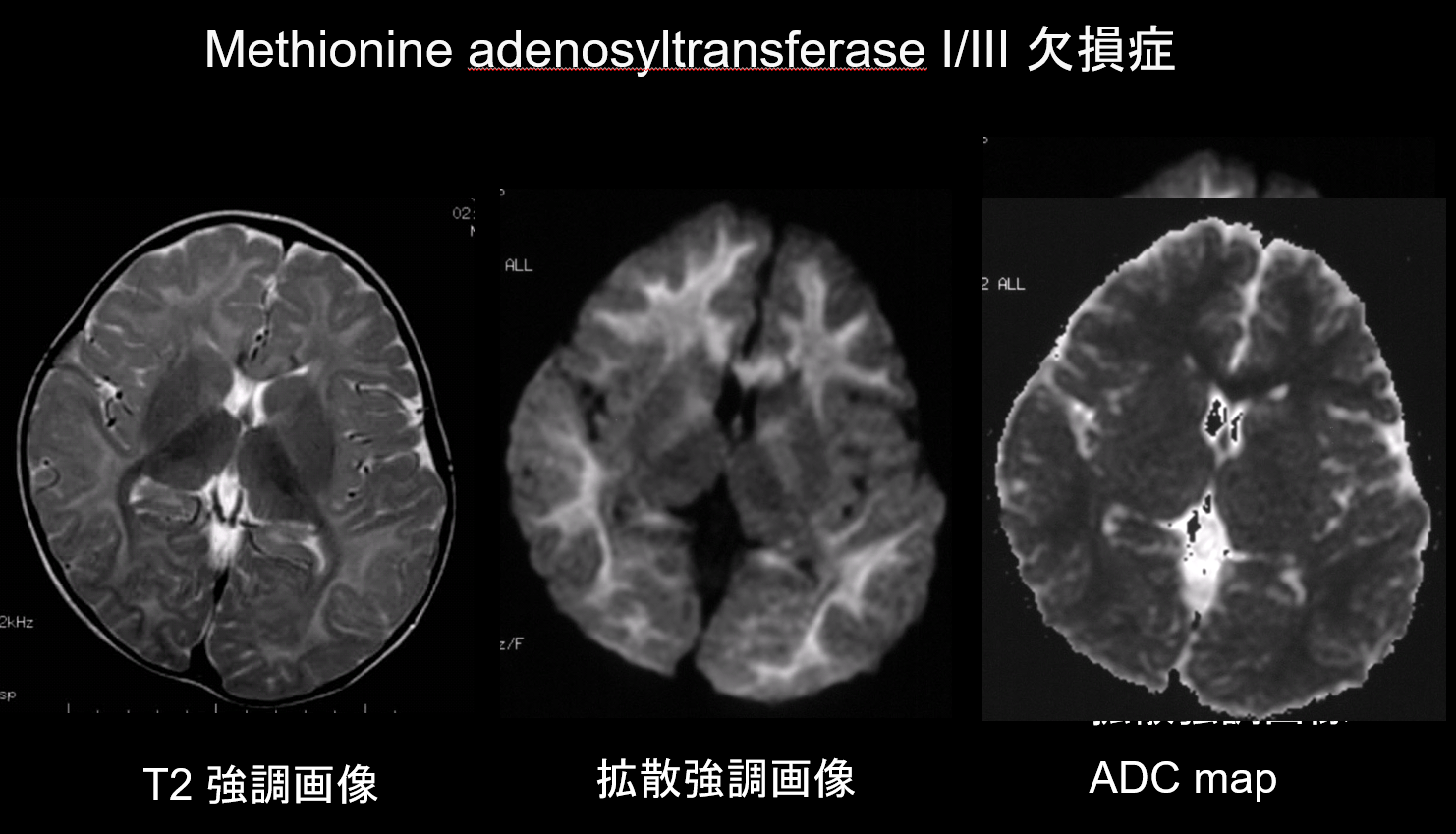
A fehérállomány-rendellenességek fő patológiái, a demielináció és a hipomielináció esetében is csökken a myelin mennyisége, ami korlátozza a diffúziót, és az extracelluláris folyadék megfelelő növekedése magas látszólagos diffúziós együtthatóval (ADC) rendelkező T2 hiperintenzitást eredményez. Ritkán fordul elő, hogy a fehérállomány-rendellenességek T2-hiperintenzitást és alacsony ADC-t is mutatnak, ezért ez a kombináció nagy diagnosztikai értékkel bír. Az intramielinikus ödéma jelenlétével jellemezhető rendellenességek a mielinhüvelyen belül és a hártyák közötti résekben alacsony ADC-t mutatnak. Ide tartozik a juharszirupos vizeletbetegség, a metionin-adenozil-transzferáz I/III hiány (9. ábra), a fenilketonúria, a nem ketotikus hiperglicinémia és a Canavan-kór. A Krabbe-kór és a metakromatikus leukodisztrófia is mutathat alacsony ADC-t egyes fehérállományi elváltozásokban, mivel a demielinizáció akut fázisában intramielines ödéma alakulhat ki.
- Van der Knaap MS, Valk J. Classification of myelin disorders. In Van der Knaap MS, Valk J, szerk. A mielinizáció és a mielin rendellenességek mágneses rezonanciája. 3. kiadás, Berlin: Springer, 2005, 20-24.
- Schiffmann R, van der Knaap MS. MRI-alapú megközelítés a fehérállomány-rendellenességek diagnosztizálásához. Neurology 2009; 72: 750-759
- Takanashi J. Diagnostic imaging of white matter disorders. Journal of the Japan Pediatric Society 2007; 111: 1243-1254.
- Van der Knaap MS, Breiter SN, Naidu S, et al. Defining and categorizing leukoencephalopathies of unknown origin: MR képalkotó megközelítés. Radiology 1999; 213: 121-133.