Diagnostic Imaging of White Matter Abnormalities | Congenital cerebral hypomyelination; Netzwerk für die Pelizaeus-Merzbacher-Krankheit und verwandte Störungen
Diagnostische Bildgebung von Anomalien der Weißen Substanz
Junichi TAKANASHI, Abteilung für Pädiatrie, Tokyo Women’s Medical University, Yachiyo Medical Center
Einführung
In diesem Beitrag wird die Vorgehensweise bei der Diagnose von Störungen, die sich in der Magnetresonanztomographie (MRT) als abnorme Signale in der weißen Substanz des Gehirns zeigen, von der Bildgebung bis zur Diagnose beschrieben. Störungen, die hauptsächlich die weiße Substanz betreffen, werden im Allgemeinen als „Leukoenzephalopathie“ oder „Störungen der weißen Substanz“ bezeichnet.1, 2) 3) Ein anderer Begriff, Leukodystrophie, wird manchmal mit Degeneration der weißen Substanz verwechselt, bezieht sich jedoch auf ein engeres Spektrum von Störungen mit einer genetischen Komponente (vererbte demyelinisierende Störungen).
Bildgebungsbasierte Klassifizierung von Störungen der weißen Substanz
Das Aufkommen der MRT hat unsere Fähigkeit, Läsionen in der weißen Substanz des zentralen Nervensystems zu erkennen, dramatisch verbessert. Viele bekannte Formen von Störungen der weißen Substanz zeigen spezifische Anzeichen in der MRT, was für ihre Diagnose nützlich ist. Die Identifizierung von Mustern von Anomalien der weißen Substanz im MRT (T1-gewichtet, T2-gewichtet oder FLAIR-Bildgebung) erleichtert die Eingrenzung der Möglichkeiten bei mehreren Differentialdiagnosen. Die Klassifizierung der Erkrankungen der weißen Substanz nach den MRT-Befunden von Schiffmann und van der Kamp ist von praktischem Wert2, 4) (Abbildung 1, Tabelle 1). Selbst wenn dies nicht zu einer endgültigen Diagnose führt, kann die Klassifizierung von Bildgebungsbefunden zur späteren Entdeckung einer neuen Störung führen. Im Folgenden beschreibe ich Störungen der weißen Substanz anhand der oben genannten MRT-Klassifikationen und erkläre die wichtigsten Arten von Störungen.
Tabelle 1. Liste der Erkrankungen nach MRT-Mustern
- Frontale Dominanz
Alexander-Krankheit, frontale Variante der X-chromatischen Adrenoleukodystrophie (ALD), metachromatische Leukodystrophie (MLD), neuroaxonale Leukodystrophie mit Sphäroiden. - Parieto-occipitale Dominanz
X-chromosomale Adrenoleukodystrophie (ALD), Morbus Krabbe, früh einsetzende peroxisomale Störungen, neonatale Hypoglykämie. - Periventrikuläre Dominanz
Metachromatische Leukodystrophie (MLD), Krabbe-Krankheit, Sjögren-Larsson-Syndrom, adulte Polyglucosankörper-Krankheit, Leukoenzephalopathie mit Hirnstamm- und Rückenmarksbeteiligung und Laktaterhöhung (LBSL), periventrikuläre Leukomalazie (PVL), HIV-Enzephalopathie, später auftretende neuronale Ceroidlipofuszinosen. - Subkortikale Dominanz
L-2-Hydroxyglutarsäureurie, Galaktosämie, Kearns-Sayer-Syndrom, Propionsäureakazie, Harnstoffzyklusstörungen, Canavan-Krankheit. - Diffuse zerebrale
Megalenzephalische Leukoenzephalopathie mit subkortikalen Zysten (MLC), Leukoenzephalopathie mit verschwindender weißer Substanz (VWM), Merosin-defiziente kongenitale Muskeldystrophie, Mitochondrienerkrankung, Molybdän-Cofaktor-Mangel, Sulfit-Oxidase-Mangel, fortgeschrittene Fälle von Störungen der weißen Substanz. - Vorherrschen oder Hervortreten der hinteren Schädelgrube
Läsionen des Kleinhirns und der Kleinhirnstiele: Zerebrotendinöse Xanthomatose (CTX), peroxisomale Störungen, Alexander-Krankheit, Leukoenzephalopathie mit Hirnstamm- und Rückenmarksbeteiligung und Laktaterhöhung (LBSL), Ahornsirup-Urin-Krankheit, Histiozytose, autosomal-dominante Leukodystrophie bei Erwachsenen im Zusammenhang mit einer Lamin-B1-Duplikation, Heroin- und Kokaintoxizität.
Hirnstammläsionen: Alexander-Krankheit, LSBL, peroxisomale Störungen, Wilson-Krankheit, adulte Polyglucosan-Krankheit, Leigh-Syndrom, Dentatorubropallidoluysian-Atrophie (DRPLA), adulte Polyglucosan-Körper-Krankheit, adulte autosomal-dominante Leukodystrophie im Zusammenhang mit einer Lamin-B1-Duplikation. - Multifokale Läsionen
TORCH-Syndrom (kongenitale Cytomegalovirus-Infektion), Brucellose, akute disseminierte Enzephalomyelitis (ADEM), Multiple Sklerose (MS), Neuromyelitis optica (NMO), zerebrale autosomal-dominante Arteriopathie mit subkortikalen Infarkten und Leukenzephalopathie (CADASIL), Atherosklerose, Amyloid-Angiopathie, COL4A1-assoziierte zerebrale Kleingefäßerkrankung, Fabry-Krankheit, Susac-Syndrom, mitochondriale Erkrankung, L-2-Hydroxyglutarsäureurie, Mukopolysaccharidose (MPS), Chromosomenanomalien (wie das 6p-Syndrom). - Läsionen mit geringer Diffusionskapazität
Ahornsirup-Urin-Krankheit, Methionin-Adenosyltransferase I/III-Mangel, Phenylketonurie, nicht-ketotische Hyperglyzinämie, Canavan-Krankheit, aktive Läsionen bei Morbus Krabbe und metachromatische Leukodystrophie.
1. Hypomyelinisierung der zerebralen weißen Substanz
Dies bezieht sich auf eine Gruppe von Erkrankungen, bei denen die Bildung der Myelinscheide gestört oder verzögert ist, und ihre Bilder ähneln denen von Neugeborenen mit unreifer Myelinisierung. Auf T2-gewichteten Bildern erscheint die weiße Substanz charakteristischerweise als weit verbreitete Hyperintensität, die im Vergleich zum Kortex schwach ausgeprägt ist. Weitere Einzelheiten finden Sie auf der Website der kongenitalen zerebralen Hypomyelinatio.
Wenn Läsionen der weißen Substanz nicht mit einer Hypomyelinisierung der zerebralen weißen Substanz übereinstimmen, muss festgestellt werden, ob sie konfluent oder multipel sind.2) Konfluente Läsionen der weißen Substanz sind in der Regel auf eine vererbte Degeneration der weißen Substanz (Leukodystrophie) zurückzuführen und sind in den meisten Fällen bilateral symmetrisch. Multiple Läsionen der weißen Substanz sind in der Regel asymmetrisch und erworben. Konfluente Läsionen der weißen Substanz werden weiter in die nachstehenden Kategorien 2-7 unterteilt.
2. Frontale Vorherrschaft
In dieser Gruppe von Erkrankungen sind ausgedehnte Läsionen der weißen Substanz vorwiegend im Frontallappen vorhanden. Dazu gehören die Alexander-Krankheit, die frontale Variante der X-chromatischen Adrenoleukodystrophie (ALD), die metachromatische Leukodystrophie (MLD) und die neuroaxonale Leukodystrophie mit Sphäroiden.
(a) Alexander-Krankheit.
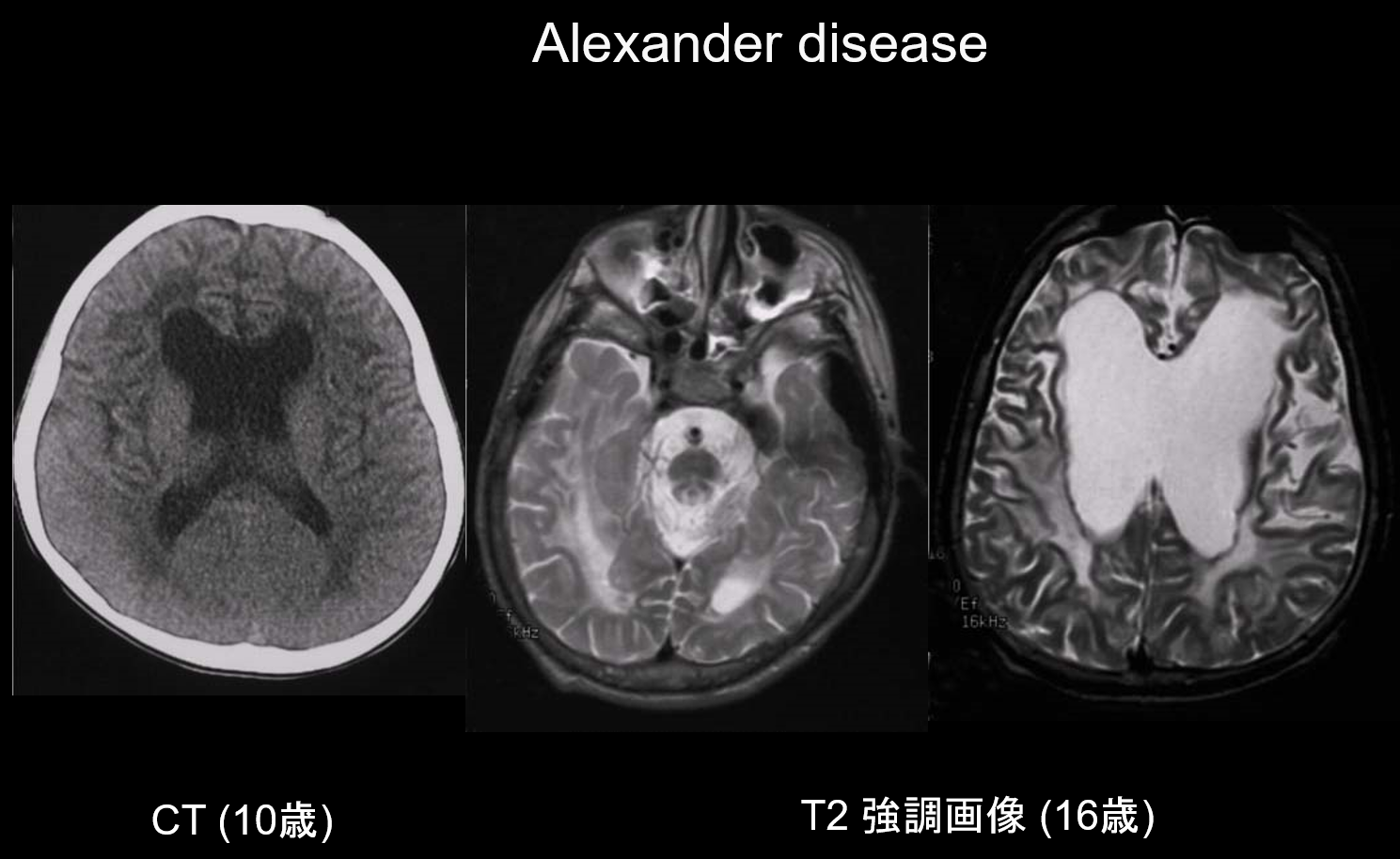
Die Alexander-Krankheit ist eine autosomal-dominant vererbte Störung, die durch eine Mutation im GFAP-Gen auf Chromosom 17q21 verursacht wird. Sie führt zu einer Anhäufung von Rosenthal-Fasern in den stellaten Gliazellen. Diese Fasern setzen sich aus GFAP und Stressproteinen (αB-Crystallin und HSP27) zusammen. Die Alexander-Krankheit tritt hauptsächlich im Säuglingsalter zwischen 3 Monaten und 2 Jahren auf und äußert sich in Megalencephalie, Entwicklungsverzögerung, spastischer Lähmung und Epilepsie. Im MRT können (i) ausgedehnte Läsionen der weißen Substanz, vorwiegend im Frontallappen, (ii) T1-hyperintense und T2-hypointense Ränder um die Seitenventrikel, (iii) Läsionen in den Basalganglien und im Thalamus, (iv) Hirnstammläsionen und (v) Kontrastverstärkung aktiver Läsionen auftreten (Abbildung 2). In den frühen Stadien treten neben Läsionen der weißen Substanz und des Putamens Schwellungen auf, die allmählich zu Atrophie oder Zystenbildung führen können.
3. Parieto-okzipitale Dominanz
Das Hauptmerkmal dieser Gruppe von Störungen sind Läsionen der parieto-okzipitalen weißen Substanz. Dazu gehören die X-chromosomale Adrenoleukodystrophie (ALD), der Morbus Krabbe, früh einsetzende peroxisomale Störungen und neonatale Hypoglykämie.
(a) Morbus Krabbe.
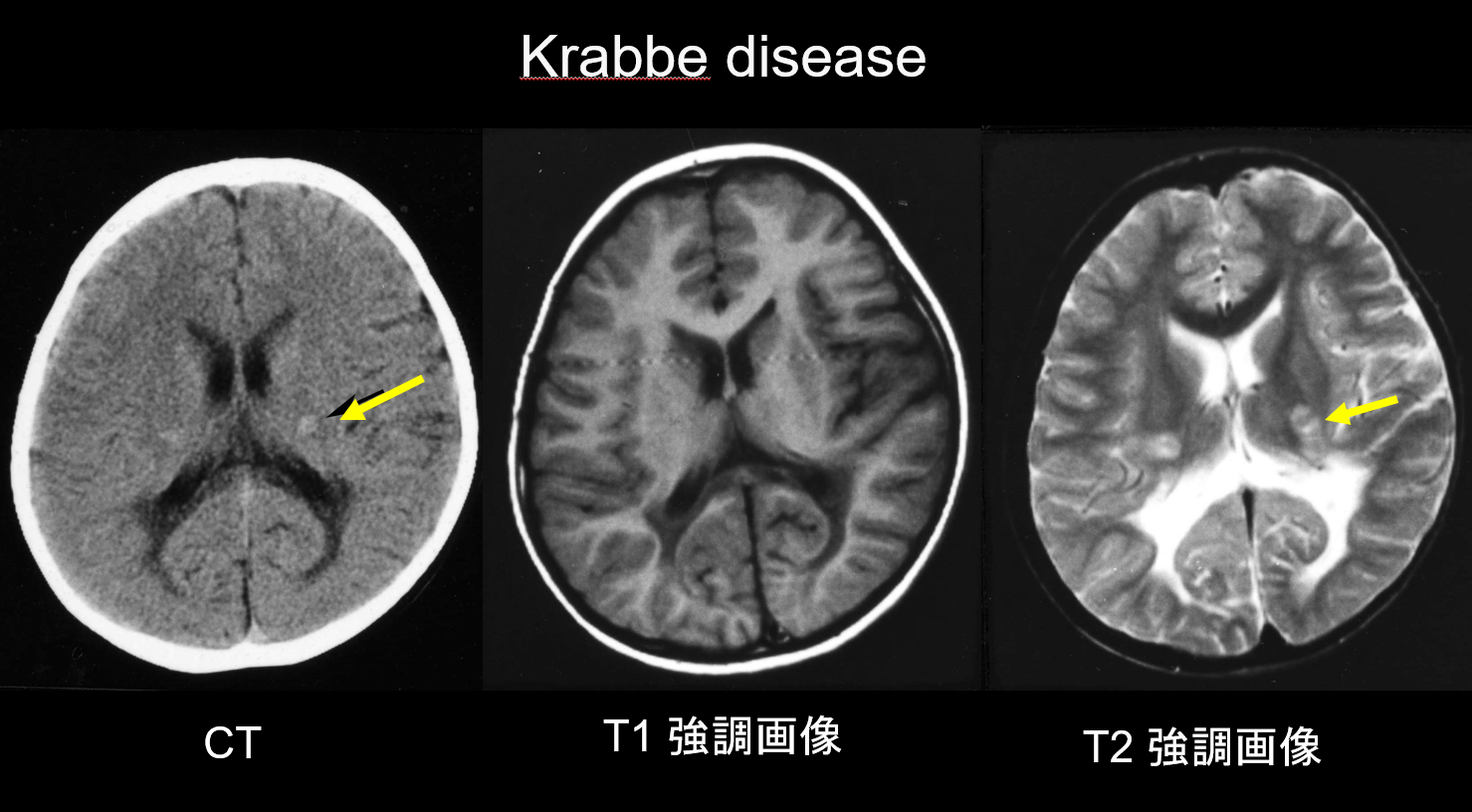
Der Morbus Krabbe ist eine autosomal rezessiv vererbte Störung (lysosomale Speicherkrankheit), die durch einen Mangel an Galaktosylceramidase (Chromosom 14q31) verursacht wird und bei der die Anhäufung von hochgradig zytotoxischem Psychosin vermutlich eine weit verbreitete Demyelinisierung verursacht. Es treten auch große, vielkernige Zellen auf, die „globoide Zellen“ genannt werden. Je nach dem Alter, in dem die Krankheit auftritt, wird sie als infantile, spät auftretende infantile, jugendliche oder erwachsene Krankheit klassifiziert. Die meisten Fälle treten im Säuglingsalter auf und beginnen mit dem Auftreten von Fieber, Reizbarkeit, Fütterungsschwierigkeiten, Entwicklungsverzögerung, peripherer Neuropathie, Spastizität und Sehnervenatrophie im Alter von 3-6 Monaten. In den frühen Stadien zeigt die Computertomographie (CT) eine charakteristische Hyperdensität im Thalamus und in der Corona radiata. Man nimmt an, dass dies auf eine hohe Dichte an globoiden Zellen und Glia-Proliferationen zurückzuführen ist. Die MRT kann auch eine T1-Hyperintensität und eine T2-Hypointensität um die Ventrikel sowie lineare Strukturen zeigen, die denen der MLD ähneln (Abbildung 3). Der Nucleus dentatus des Kleinhirns, die weiße Substanz des Kleinhirns und die Pyramidenbahn des Hirnstamms weisen bereits in einem frühen Stadium eine T2-Hyperintensität auf.
(b) X-chromosomale Adrenoleukodystrophie
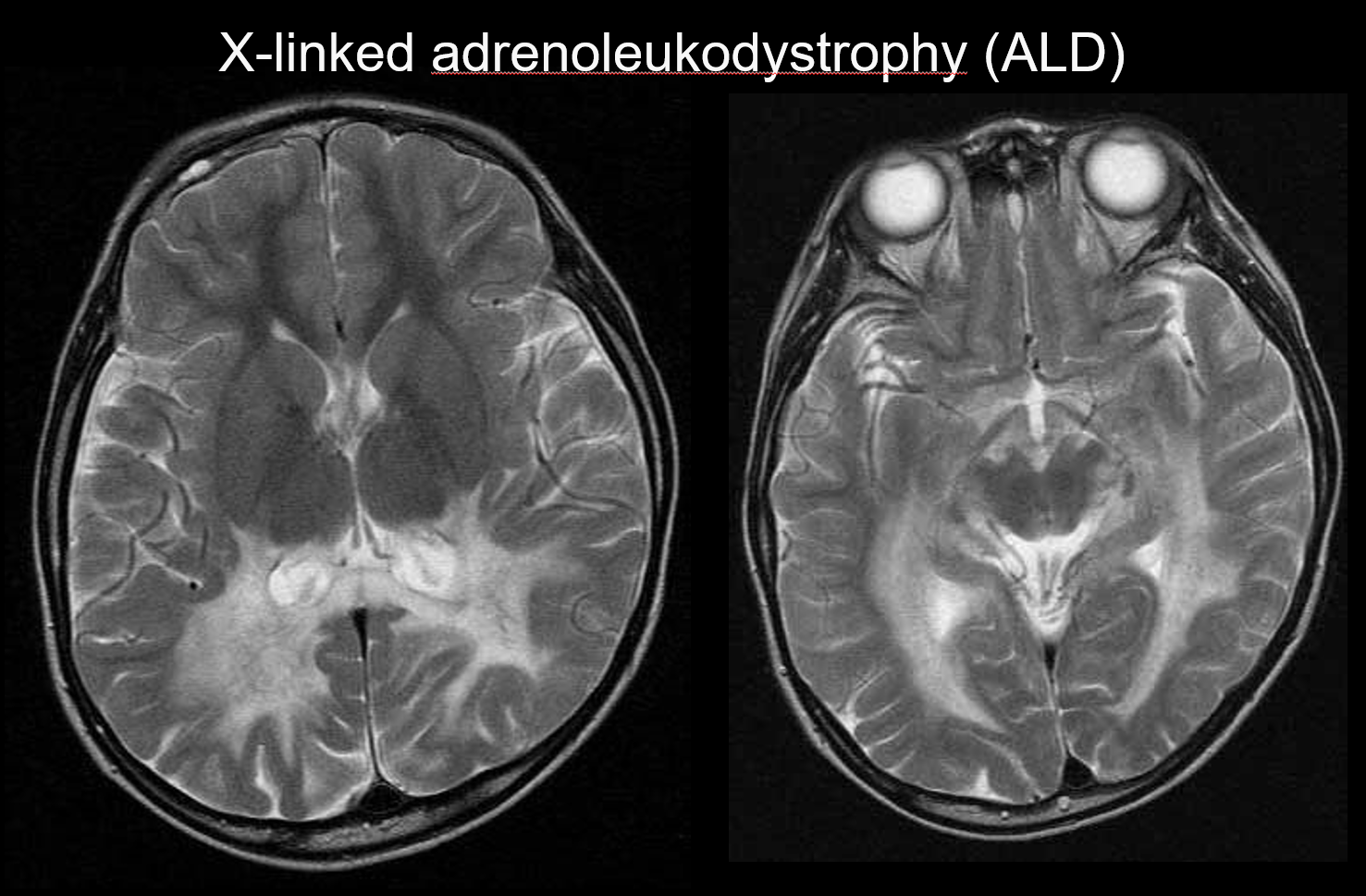
Die X-chromosomale Adrenoleukodystrophie (ALD) ist eine X-chromosomal rezessiv vererbte Störung (peroxisomale Störung), die durch eine Anomalie des ABCD1-Gens (Chromosom Xq28) verursacht wird. Eine gestörte β-Oxidation führt zu einer Anhäufung von sehr langkettigen Fettsäuren in der weißen Hirnsubstanz und den Nebennieren, was zu Demyelinisierung und Nebenniereninsuffizienz führt. Die ALD wird in die zerebrale Form im Kindes-, Jugend- und Erwachsenenalter, die Adrenomyeloneuropathie (AMN) und die reine Addison-Krankheit eingeteilt. Die zerebrale Form im Kindesalter entwickelt sich im Alter von 5 bis 8 Jahren, wobei Symptome wie intellektueller Verfall, spastischer Gang sowie Seh- und Hörstörungen auftreten. Pathologisch gesehen schreitet die Demyelinisierung von der weißen Substanz, die den Trigonus des Seitenventrikels umgibt, zum Splenium des Corpus callosum fort und dehnt sich allmählich nach anterolateral aus. Entsprechend der Krankheitspathologie zeigen sich im MRT symmetrische T2-Hyperintensitäten und T1-Hypointensitäten, die sich anterolateral von der weißen Substanz um das Trigon des Seitenventrikels erstrecken, wobei an den Rändern eine Kontrastmittelanreicherung erkennbar ist (Abbildung 4). Auch Läsionen des Corticospinaltrakts sind erkennbar.
4. Periventrikuläre Vorherrschaft
Diese Erkrankungen sind hauptsächlich durch Läsionen in der weißen Substanz gekennzeichnet, die die lateralen Ventrikel umgibt, wobei die subkortikale weiße Substanz (U-Fasern) erhalten bleibt. Dieses Muster tritt bei zahlreichen Erkrankungen auf, einschließlich MLD, und ist daher relativ unspezifisch. Leicht abnorme Signale um die Seitenventrikel werden auch bei kortikaler Degeneration beobachtet, insbesondere bei neuronalen Ceroidlipofuszinosen, die sich nach dem Säuglingsalter entwickeln.
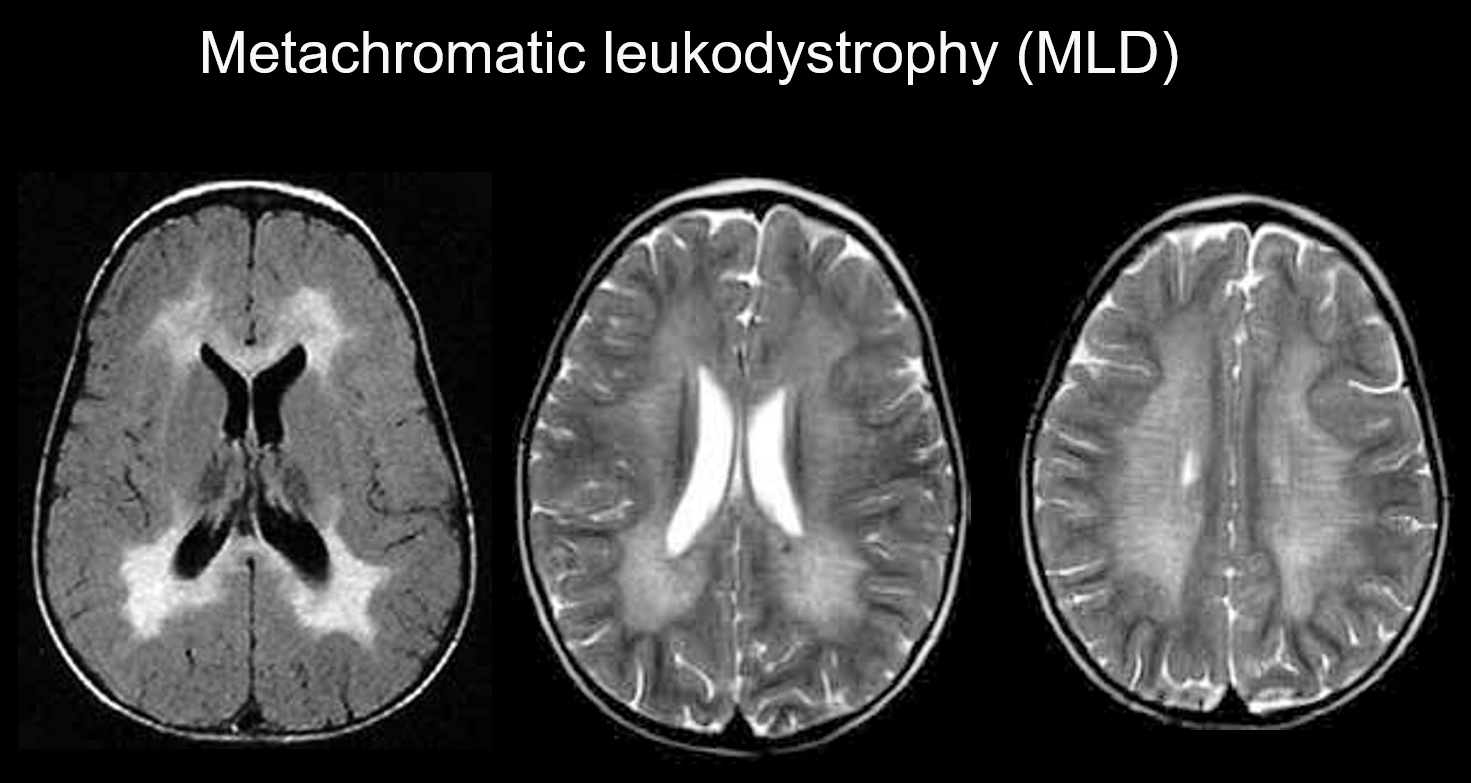
(a) Metachromatische Leukodystrophie.
Die metachromatische Leukodystrophie ist eine autosomal rezessiv vererbte Störung (lysosomale Speicherkrankheit), die durch einen Arylsulfatase-A-Mangel (Chromosom 22q13.31) verursacht wird und bei der die Anhäufung von hochtoxischem Sulfatid zu einer Demyelinisierung führt. Je nach dem Alter, in dem die Krankheit auftritt, wird sie als kongenital, im Kindesalter, im Jugendalter oder im Erwachsenenalter auftretende Krankheit klassifiziert. Zu den Symptomen gehören kognitive Regression, spastische Lähmungen, unwillkürliche Bewegungen, periphere Neuropathie und Atrophie des Sehnervs. Die Erkrankung zeigt sich in der T2-gewichteten Bildgebung als Hyperintensitäten der weißen Substanz, vor allem um die Seitenventrikel, und in der T1-gewichteten Bildgebung als leichte Hypointensitäten. Die Läsionen befinden sich in der Regel vorwiegend im Frontallappen. Innerhalb der weit verbreiteten abnormalen Signale in der weißen Substanz können Bänder mit normaler Intensität (Tigerstreifen) zu sehen sein (Abbildung 5). Man nimmt an, dass diese auf die teilweise Erhaltung der Myelinscheide im perivaskulären Raum und auf die Ansammlung von Abbauprodukten der Myelinscheide in Makrophagen zurückzuführen sind.
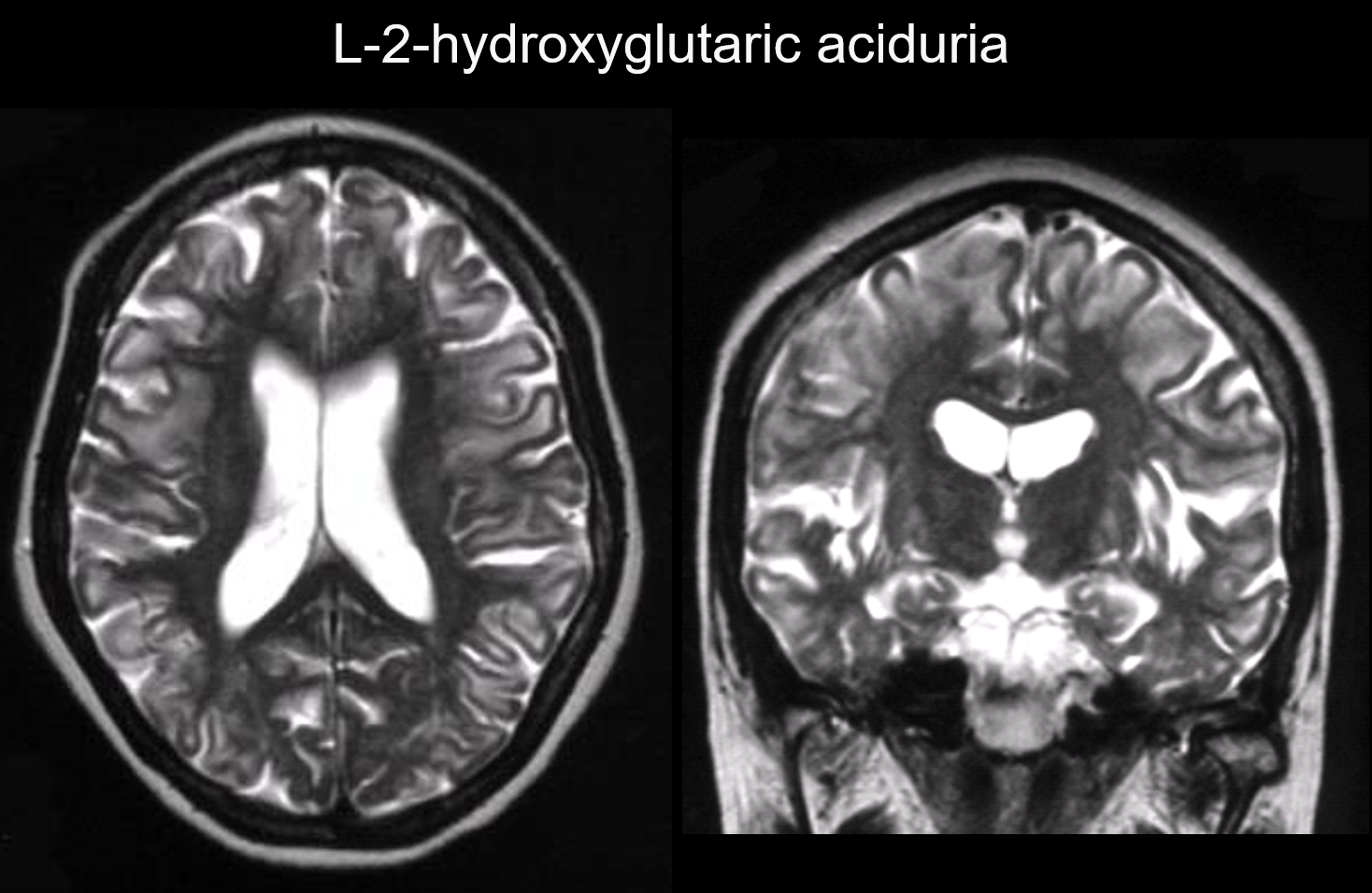
5. Subkortikale Vorherrschaft
Bei diesen Störungen treten Läsionen hauptsächlich in der subkortikalen weißen Substanz auf, einschließlich der U-Fasern. Zu den Erkrankungen mit diesem Muster gehören die L-2-Hydroxyglutarsäureurie (Abbildung 6), Galaktosämie, das Kearns-Sayer-Syndrom, Propionsäureakazie, Harnstoffzyklusstörungen und die Canavan-Krankheit im Frühstadium.
6. Diffuse zerebrale
Bei diesen Erkrankungen treten abnorme Signale in der gesamten weißen Substanz des Gehirns auf. Sie weisen starke T2-Hyperintensitäten im Vergleich zu den T2-Signalen der nicht myelinisierten weißen Substanz auf (Hypomyelinisierung). Neben Fällen von megalencephaler Leukoenzephalopathie mit subkortikalen Zysten und Leukoenzephalopathie mit verschwindender weißer Substanz zeigen Patienten mit jeder Art von Störung der weißen Substanz mit fortschreitender Erkrankung dieses Muster.
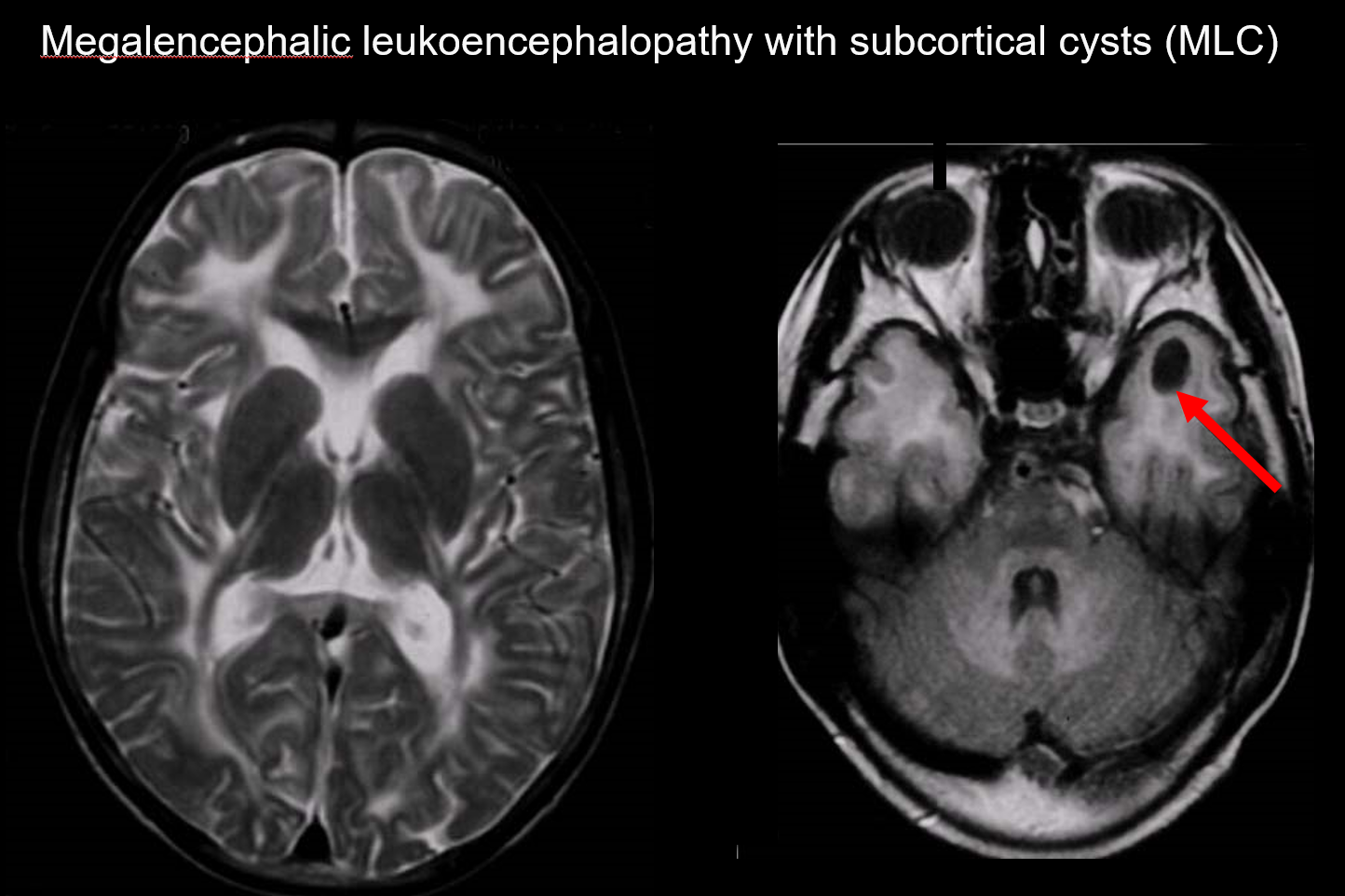
(a) Megalencephale Leukoenzephalopathie mit subkortikalen Zysten (MLC)
MLC ist eine autosomal rezessiv vererbte Erkrankung, die durch eine Anomalie des MLC1-Gens verursacht wird und deren Beginn im Säuglingsalter durch Megalozephalie, langsam fortschreitende motorische Verschlechterung, Ataxie und Spastik gekennzeichnet ist. Die MRT zeigt charakteristische, weit verbreitete abnorme Signale in der weißen Substanz und eine leichte Schwellung der weißen Substanz sowie eine Zystenbildung im Parietal- und Temporallappen (Abbildung 7).7, 8) Die T1- und T2-gewichtete Bildgebung zeigt abnorme weiße Substanz, während die Zysten alle eine T1-Hypointensität und T2-Hyperintensität aufweisen, wodurch sie besonders schwer zu erkennen sind. Die FLAIR-Bildgebung, die Zysten (Wasser) als Hypointensitäten sichtbar macht, ist für die Diagnose von großem Nutzen. Sie ist bei Japanern häufiger als die vacuolating megalencephalic leukoencephalopathy.
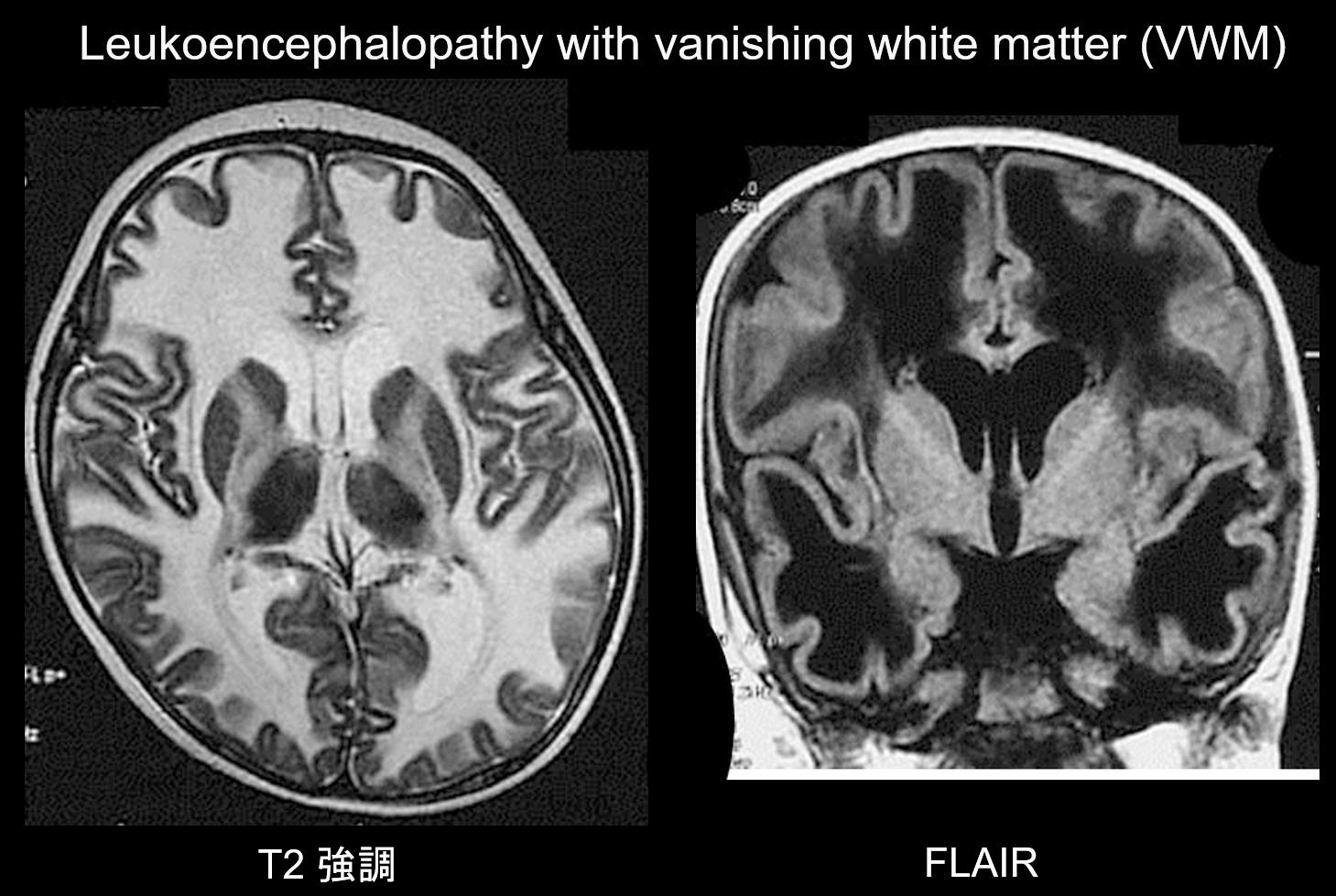
(b) Leukoenzephalopathie mit verschwindender weißer Substanz.
Leukoenzephalopathie mit verschwindender weißer Substanz (VWM) ist eine autosomal rezessiv vererbte Erkrankung, die durch einen Mangel an eIF2B verursacht wird, einem mit eIF2 assoziierten Protein, das Initiator-tRNA auf Ribosomen überträgt. eIF2B besteht aus fünf verschiedenen Proteinen, die alle unterschiedliche genetische Loci haben. VWM ist nachweislich dieselbe Störung wie die zerebelläre Ataxie im Kindesalter und die zentrale Hypomyelinisierung (CACH). Die Patienten sind während der Neugeborenenperiode und im frühen Säuglingsalter normal, aber nach dem Auftreten der Krankheit (gewöhnlich im Alter von 2-6 Jahren) entwickeln sie eine langsam fortschreitende kognitive Regression, Spastizität und Ataxie. Es ist bekannt, dass sich diese Symptome durch Infektionen oder leichte Traumata verschlimmern. Die zerebrale weiße Substanz weist eine weit verbreitete T2-Hyperintensität und T1-Hypointensität auf und wird im Laufe der Zeit allmählich durch Flüssigkeit ersetzt (wie der Name schon sagt, verschwindet die weiße Substanz) (Abbildung 8). Die zystische weiße Substanz enthält gebänderte Strukturen, von denen man annimmt, dass sie das verbleibende Gewebe darstellen. Abnormale Signale sind auch im Hirnstamm zu sehen, insbesondere im zentralen Tegmentaltrakt. Die FLAIR-Bildgebung ist für die Diagnose dieser Störung von großem Nutzen.
7. Überwiegender Anteil der hinteren Schädelgrube oder Vorwölbung
Diese Störungen sind durch Läsionen vorwiegend im Hirnstamm und im Kleinhirn gekennzeichnet. Läsionen der zerebellären weißen Substanz können durch Erkrankungen wie zerebrotendinöse Xanthomatose (CTX), peroxisomale Störungen, Alexander-Krankheit, Leukoenzephalopathie mit Hirnstamm- und Rückenmarksbeteiligung und Laktaterhöhung (LBSL), Ahornsirup-Urin-Krankheit, Histiozytose sowie Heroin- und Kokaintoxizität verursacht werden. Hirnstammläsionen können durch Erkrankungen wie die Alexander-Krankheit, LSBL und die adulte Polyglucosan-Krankheit verursacht werden. Läsionen des mittleren Kleinhirnstamms treten beim fragilen X-Syndrom und bei der autosomal-dominanten Leukodystrophie bei Erwachsenen auf, die mit einer Lamin-B1-Duplikation zusammenhängt.
8. Multifokale Läsionen
Im Gegensatz zu den unter 2-7 beschriebenen konfluenten Läsionen führen die in diesem Abschnitt beschriebenen Störungen zu multifokalen (verstreuten) Läsionen. Dazu gehören Infektionen wie das TORCH-Syndrom (aufgrund einer kongenitalen Cytomegalovirus-Infektion oder einer anderen Ursache) und Brucellose; entzündliche Erkrankungen wie die akute disseminierte Enzephalomyelitis (ADEM), Multiple Sklerose (MS) und Neuromyelitis optica (NMO); Vaskulopathien wie zerebrale autosomal-dominante Arteriopathie mit subkortikalen Infarkten und Leukoenzephalopathie (CADASIL), Atherosklerose, Amyloidangiopathie, COL4A1-assoziierte zerebrale Kleingefäßerkrankung, Morbus Fabry und Susac-Syndrom; und vererbte Erkrankungen wie mitochondriale Erkrankungen, L-2-Hydroxyglutarsäureurie, Mukopolysaccharidose (MPS) und Chromosomenanomalien (wie das 6p-Syndrom).
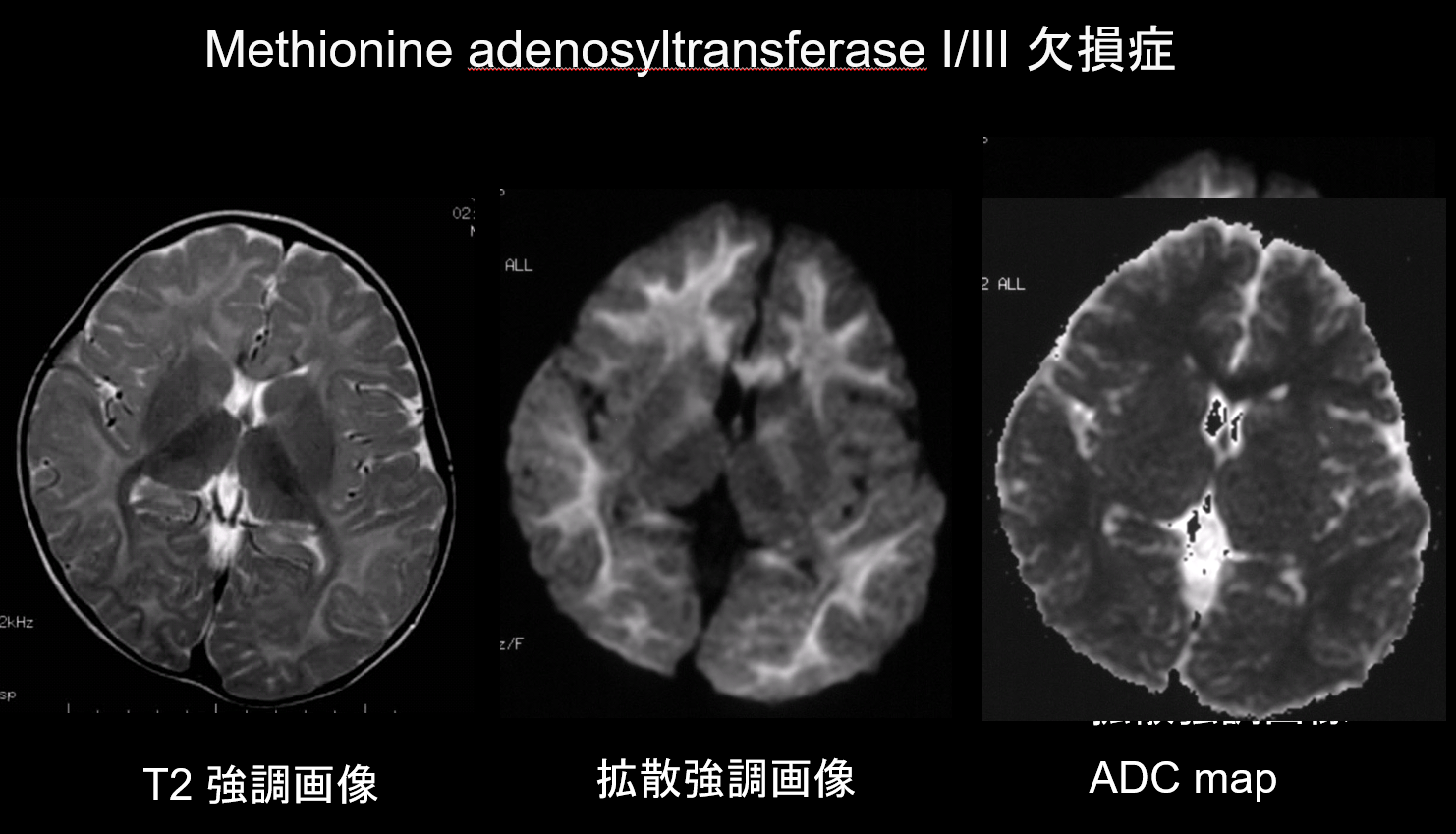
9. Läsionen mit geringer Diffusionskapazität
Sowohl bei der Demyelinisierung als auch bei der Hypomyelinisierung, den Hauptpathologien der Erkrankungen der weißen Substanz, kommt es zu einer Abnahme der Myelinmenge, was die Diffusion einschränkt, und die entsprechende Zunahme der extrazellulären Flüssigkeit führt zu T2-Hyperintensitäten mit einem hohen scheinbaren Diffusionskoeffizienten (ADC). Es ist selten, dass Störungen der weißen Substanz sowohl T2-Hyperintensitäten als auch einen niedrigen ADC-Wert aufweisen, und diese Kombination ist daher von hohem diagnostischem Wert. Störungen, die durch das Vorhandensein eines intramyelinischen Ödems innerhalb der Myelinscheide und in den Lücken zwischen den Hüllen gekennzeichnet sind, weisen eine niedrige ADC auf. Dazu gehören die Ahornsirup-Urin-Krankheit, der Methionin-Adenosyltransferase-I/III-Mangel (Abbildung 9), die Phenylketonurie, die nicht-ketotische Hyperglycinämie und die Canavan-Krankheit. Die Krabbe-Krankheit und die metachromatische Leukodystrophie können in einigen Läsionen der weißen Substanz ebenfalls eine niedrige ADC aufweisen, da während der akuten Phase der Demyelinisierung ein intramyelinisches Ödem auftreten kann.
- Van der Knaap MS, Valk J. Classification of myelin disorders. In Van der Knaap MS, Valk J, eds. Magnetische Resonanz der Myelinisierung und Myelinstörungen. 3rd ed. Berlin: Springer, 2005, 20-24.
- Schiffmann R, van der Knaap MS. Ein MRT-basierter Ansatz für die Diagnose von Störungen der weißen Substanz. Neurology 2009; 72: 750-759
- Takanashi J. Diagnostic imaging of white matter disorders. Journal of the Japan Pediatric Society 2007; 111: 1243-1254.
- Van der Knaap MS, Breiter SN, Naidu S, et al. Defining and categorizing leukoencephalopathies of unknown origin: MR imaging approach. Radiology 1999; 213: 121-133.