Diagnostické zobrazování abnormalit bílé hmoty | Vrozená hypomyelinizace mozku; síť pro Pelizaeus-Merzbacherovu chorobu a příbuzné poruchy
Diagnostické zobrazování abnormalit bílé hmoty
Junichi TAKANASHI, oddělení pediatrie, Tokijská ženská lékařská univerzita, Yachiyo Medical Center
Úvod
V tomto článku uvádím přístup používaný při diagnostice poruch, které se projevují jako abnormální signály v bílé hmotě mozkové na magnetické rezonanci (MRI), od zobrazování až po diagnostiku. Poruchy, které postihují hlavně bílou hmotu, se obecně označují jako „leukoencefalopatie“ nebo „poruchy bílé hmoty“ v angličtině. 1. Jaké poruchy se vyskytují v bílé hmotě? 2) 3) Jiný termín, leukodystrofie, se někdy zaměňuje s degenerací bílé hmoty, ale ten ve skutečnosti označuje užší spektrum poruch s genetickou složkou (dědičné demyelinizační poruchy).
Klasifikace poruch bílé hmoty založená na zobrazování
Příchod magnetické rezonance dramaticky zlepšil naši schopnost detekovat léze v bílé hmotě centrálního nervového systému. Mnoho známých forem poruch bílé hmoty vykazuje specifické znaky na MRI, což je užitečné pro jejich diagnostiku. Identifikace vzorců abnormalit bílé hmoty pozorovaných na MRI (T1 vážené, T2 vážené nebo FLAIR zobrazení) usnadňuje zúžení možností při více diferenciálních diagnózách. Praktický význam má Schiffmannova a van der Kampova klasifikace poruch bílé hmoty podle nálezů na MRI2, 4) (obrázek 1, tabulka 1). I když nevede ke konečné diagnóze, může klasifikace zobrazovacích nálezů vést k pozdějšímu objevu nové poruchy. Zde popisuji poruchy bílé hmoty z hlediska výše uvedených klasifikací MRI a vysvětluji hlavní typy poruch.
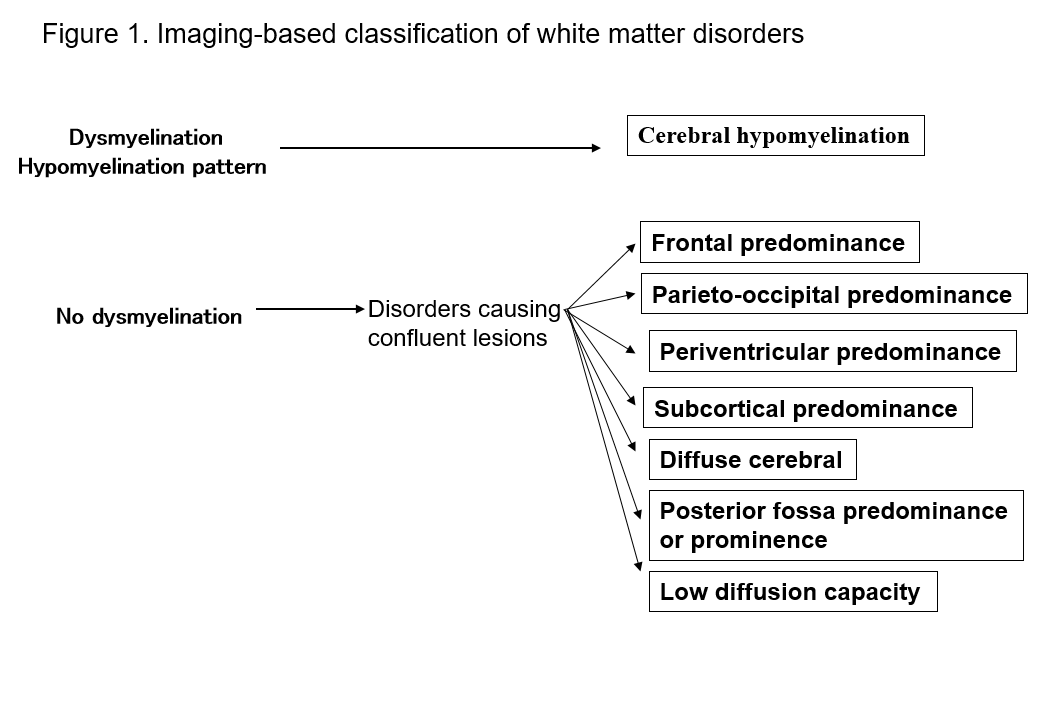
Tabulka 1. Seznam poruch podle vzorů MRI
- Frontální predominance
Alexanderova choroba, frontální varianta X-vázané adrenoleukodystrofie (ALD), metachromatická leukodystrofie (MLD), neuroaxiální leukodystrofie se sféroidy. - Parieto-okcipitální predominance
X-vázaná adrenoleukodystrofie (ALD), Krabbeho nemoc,časný nástup peroxizomálních poruch, novorozenecká hypoglykémie. - Periventrikulární predominance
Metachromatická leukodystrofie (MLD), Krabbeho nemoc, Sjögrenův-Larssonův syndrom, polyglukosanové tělísko u dospělých, leukoencefalopatie s postižením mozkového kmene a míchy a elevací laktátu (LBSL), periventrikulární leukomalacie (PVL), HIV encefalopatie, neuronální ceroidní lipofuscinózy s pozdějším nástupem. - Subkortikální predominance
L-2-hydroxyglutarová acidurie, galaktosemie, Kearns-Sayerův syndrom, propionová akademie, poruchy močovinového cyklu, Canavanova choroba. - Difúzní mozková
Megalencefalická leukoencefalopatie se subkortikálními cystami (MLC), leukoencefalopatie se zánikem bílé hmoty (VWM), vrozená svalová dystrofie s nedostatkem merosinu, mitochondriální onemocnění, nedostatek molybdenového kofaktoru, nedostatek sulfit oxidázy, pokročilé případy poruch bílé hmoty. - Převaha nebo prominence zadní jámy
Léze mozečku a mozečkových stopek: cerebrotendinózní xantomatóza (CTX), peroxizomální poruchy, Alexandrova nemoc, leukoencefalopatie s postižením mozkového kmene a míchy a elevací laktátu (LBSL), nemoc javorového sirupu v moči, histiocytóza, autozomálně dominantní leukodystrofie dospělých související s duplikací lamin B1, toxicita heroinu a kokainu.
Postižení mozkového kmene: Alexandrova choroba, LSBL, peroxizomální poruchy, Wilsonova choroba, polyglukosanová choroba dospělých, Leighův syndrom, dentatorubropallidoluysanová atrofie (DRPLA), polyglukosanová choroba dospělých, autozomálně dominantní leukodystrofie dospělých související s duplikací lamin B1. - Multifokální léze
TORCH syndrom (vrozená cytomegalovirová infekce), brucelóza, akutní diseminovaná encefalomyelitida (ADEM), roztroušená skleróza (RS), neuromyelitis optica (NMO), mozková autozomálně dominantní arteriopatie se subkortikálními infarkty a leukoencefalopatií (CADASIL), ateroskleróza, amyloidní angiopatie, mozkové onemocnění malých cév spojené s COL4A1, Fabryho choroba, Susacův syndrom, mitochondriální onemocnění, L-2-hydroxyglutarová acidurie, mukopolysacharidóza (MPS), chromozomální abnormality (např. 6p-syndrom). - Léze s nízkou difuzní kapacitou
Maple syrup urine disease, deficit methionin adenosyltransferázy I/III, fenylketonurie, neketotická hyperglycinemie, Canavanova nemoc, aktivní léze u Krabbeho nemoci a metachromatická leukodystrofie.
1. Hypomyelinizace bílé hmoty mozkové
Jedná se o skupinu poruch, u nichž je porušena nebo opožděna tvorba myelinové pochvy a její obraz se podobá obrazu novorozenců s nezralou myelinizací. Na T2 vážených snímcích se bílá hmota charakteristicky jeví jako rozsáhlá hyperintenzita, která je ve srovnání s kůrou slabá. Podrobnější informace naleznete na stránce vrozená hypomyelinizace mozku.
Pokud léze bílé hmoty neodpovídají hypomyelinizaci bílé hmoty mozku, je třeba určit, zda jsou splývající nebo mnohočetné.2) Splývající léze bílé hmoty jsou obvykle způsobeny dědičnou degenerací bílé hmoty (leukodystrofií) a ve většině případů jsou bilaterálně symetrické. Mnohočetné léze bílé hmoty jsou obvykle asymetrické a získané. Konfluentní léze bílé hmoty se dále dělí do níže uvedených kategorií 2-7.
2. Frontální predominance
V této skupině poruch jsou rozsáhlé léze bílé hmoty přítomny převážně ve frontálním laloku. Patří sem Alexandrova nemoc, frontální varianta adrenoleukodystrofie vázané na chromozom X (ALD), metachromatická leukodystrofie (MLD) a neuroaxiální leukodystrofie se sféroidy.
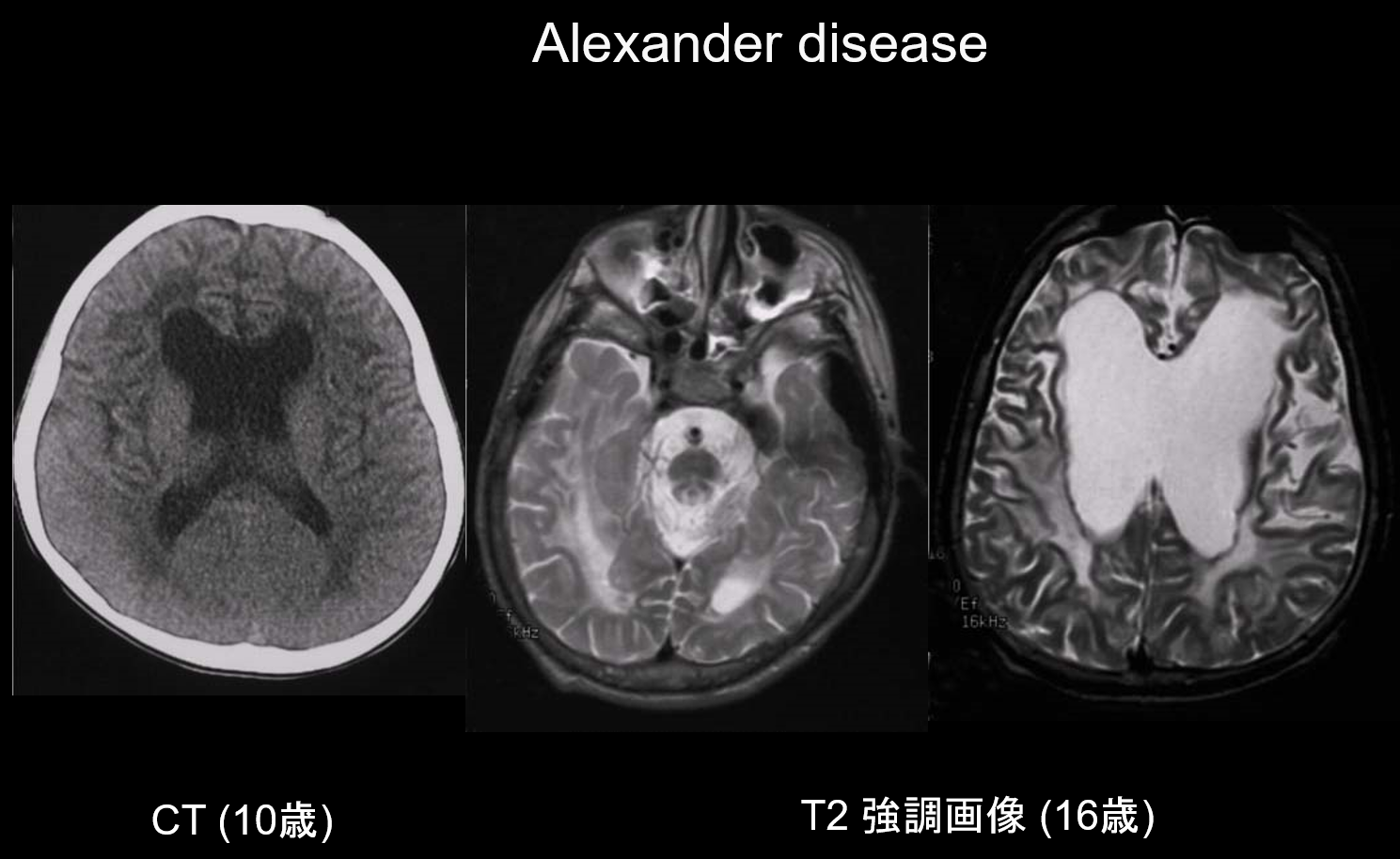
(a) Alexandrova nemoc.
Alexandrova nemoc je autozomálně dominantně dědičné onemocnění způsobené mutací v genu GFAP na chromozomu 17q21. Má za následek hromadění Rosenthalových vláken ve hvězdicovitých gliových buňkách. Tato vlákna jsou tvořena GFAP a stresovými proteiny (αB-krystalin a HSP27. Alexandrova choroba se vyskytuje především v kojeneckém věku, mezi 3 měsíci a 2 lety, s výskytem megalencefalie, vývojové retardace, spastické obrny a epilepsie. Na magnetické rezonanci se mohou objevit (i) rozsáhlé léze bílé hmoty, převážně ve frontálním laloku; (ii) T1 hyperintenzní a T2 hypointenzní ohraničení kolem postranních komor; (iii) léze v bazálních gangliích a thalamu; (iv) léze v mozkovém kmeni a (v) kontrastní zesílení aktivních lézí (obrázek 2). V časných stadiích je spolu s lézemi bílé hmoty a putamen patrný otok, který může postupně způsobit atrofii nebo tvorbu cyst.
3. Parieto-okcipitální predominance
Hlavním znakem této skupiny poruch je parieto-okcipitální léze bílé hmoty. Patří sem adrenoleukodystrofie vázaná na chromozom X (ALD), Krabbeho nemoc, peroxizomální poruchy s časným nástupem a novorozenecká hypoglykémie.
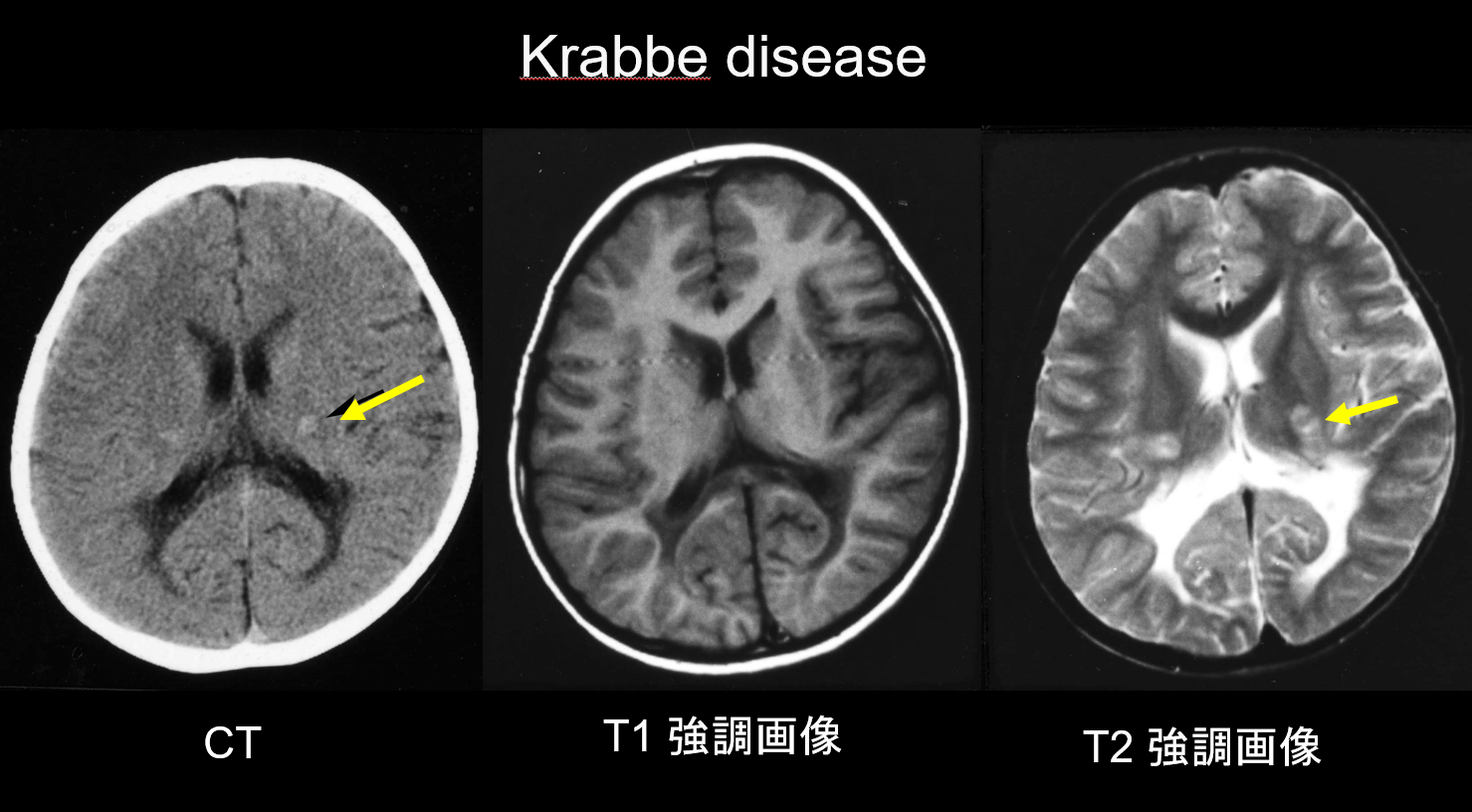
(a) Krabbeho nemoc.
Krabbeho nemoc je autozomálně recesivně dědičné onemocnění (lysozomální střádavá choroba) způsobené nedostatkem galaktosylceramidázy (chromozom 14q31), u kterého se předpokládá, že akumulace vysoce cytotoxického psychosinu způsobuje rozsáhlou demyelinizaci. Objevují se také velké vícejaderné buňky zvané „globoidní buňky“. V závislosti na věku, ve kterém se objeví, se klasifikuje jako infantilní, pozdní infantilní, juvenilní nebo dospělé onemocnění. Většina případů je kojenecká a začíná výskytem horečky, podrážděnosti, potíží s krmením, vývojovou retardací, periferní neuropatií, spasticitou a atrofií zrakového nervu ve věku 3-6 měsíců. V časných stadiích odhalí počítačová tomografie (CT) charakteristickou hyperdenzitu v thalamu a corona radiata. Předpokládá se, že odráží vysokou hustotu globoidních buněk a proliferaci glií. MRI může také ukázat hyperintenzitu T1 a hypointenzitu T2 v okolí komor a lineární struktury podobné těm, které se vyskytují u MLD (obrázek 3). Dentátní jádro mozečku, bílá hmota mozečku a pyramidový trakt mozkového kmene vykazují hyperintenzitu T2 již od raného stadia
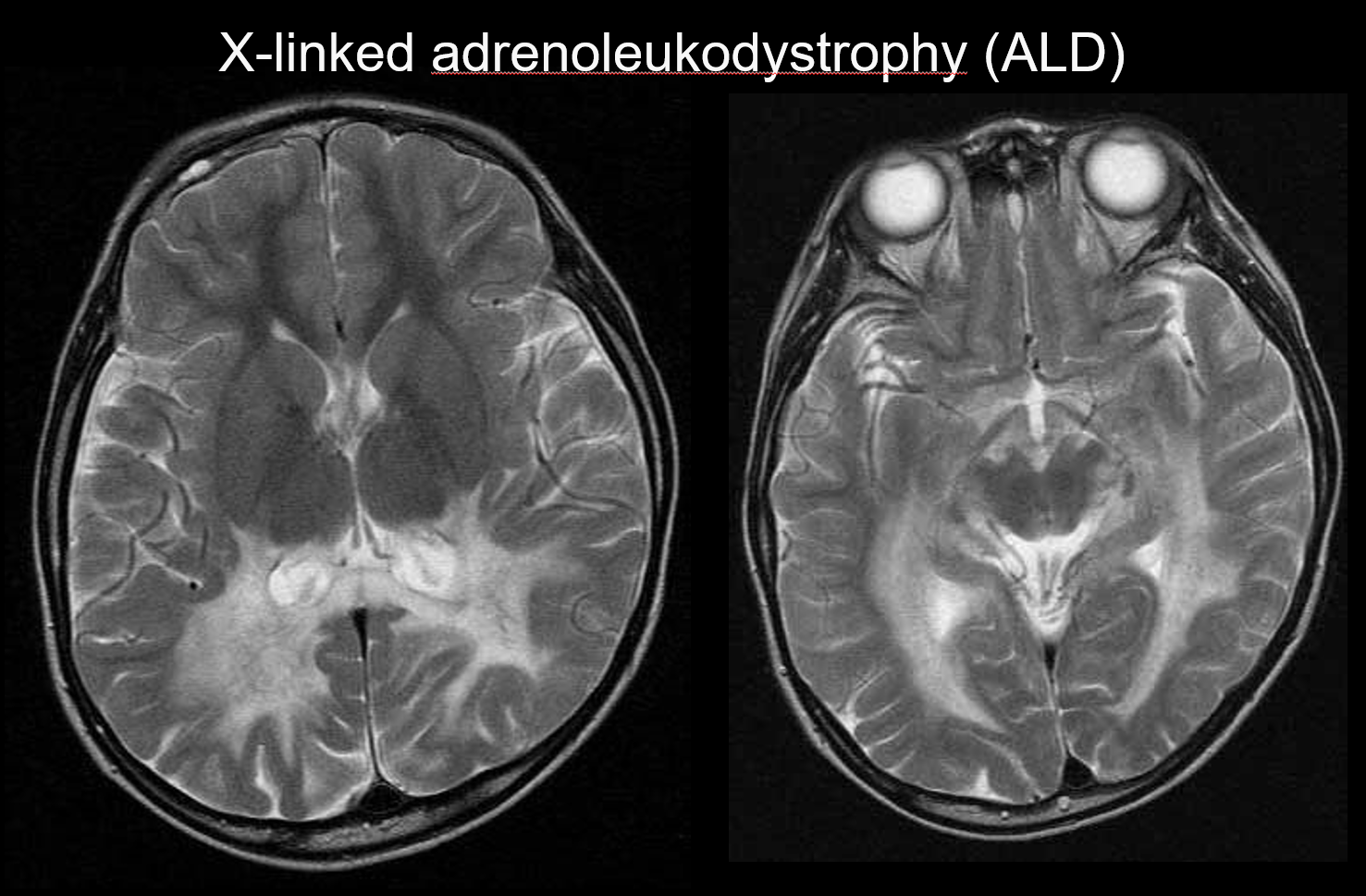
(b) X-vázaná adrenoleukodystrofie
X-vázaná adrenoleukodystrofie (ALD) je X-vázaná recesivní dědičná porucha (peroxizomální porucha) způsobená abnormalitou genu ABCD1 (chromozom Xq28). Porucha β-oxidace vede k hromadění mastných kyselin s velmi dlouhým řetězcem v bílé hmotě mozku a nadledvinách, což způsobuje demyelinizaci a nedostatečnost nadledvin. ALD se dělí na dětskou, adolescentní a dospělou mozkovou formu, adrenomyeloneuropatii (AMN) a pouze Addisonovu nemoc. Dětská cerebrální forma se rozvíjí ve věku 5-8 let, přičemž se objevují příznaky zahrnující zhoršení intelektu, spastickou chůzi a poruchy zraku a sluchu. Patologicky demyelinizace postupuje od bílé hmoty obklopující trigon postranní komory ke splenium corpus callosum a postupně se rozšiřuje anterolaterálně. Jako odraz patologie onemocnění jsou na MRI patrné symetrické hyperintenzity T2 a hypointenzity T1, které se rozšiřují anterolaterálně od bílé hmoty obklopující trigon postranní komory, s kontrastním zesílením patrným na okrajích (obrázek 4). Patrné jsou rovněž léze kortikospinálního traktu.
4. Periventrikulární predominance
Tyto poruchy jsou charakterizovány především lézemi v bílé hmotě obklopující postranní komory, přičemž subkortikální bílá hmota (U-vlákna) je zachována. Tento vzorec se vyskytuje u mnoha poruch, včetně MLD, a je tedy poměrně nespecifický. Mírně abnormální signály kolem postranních komor se vyskytují také u kortikální degenerace, zejména u neuronálních ceroidních lipofuscinóz, které se vyvíjejí po narození.
(a) Metachromatická leukodystrofie.
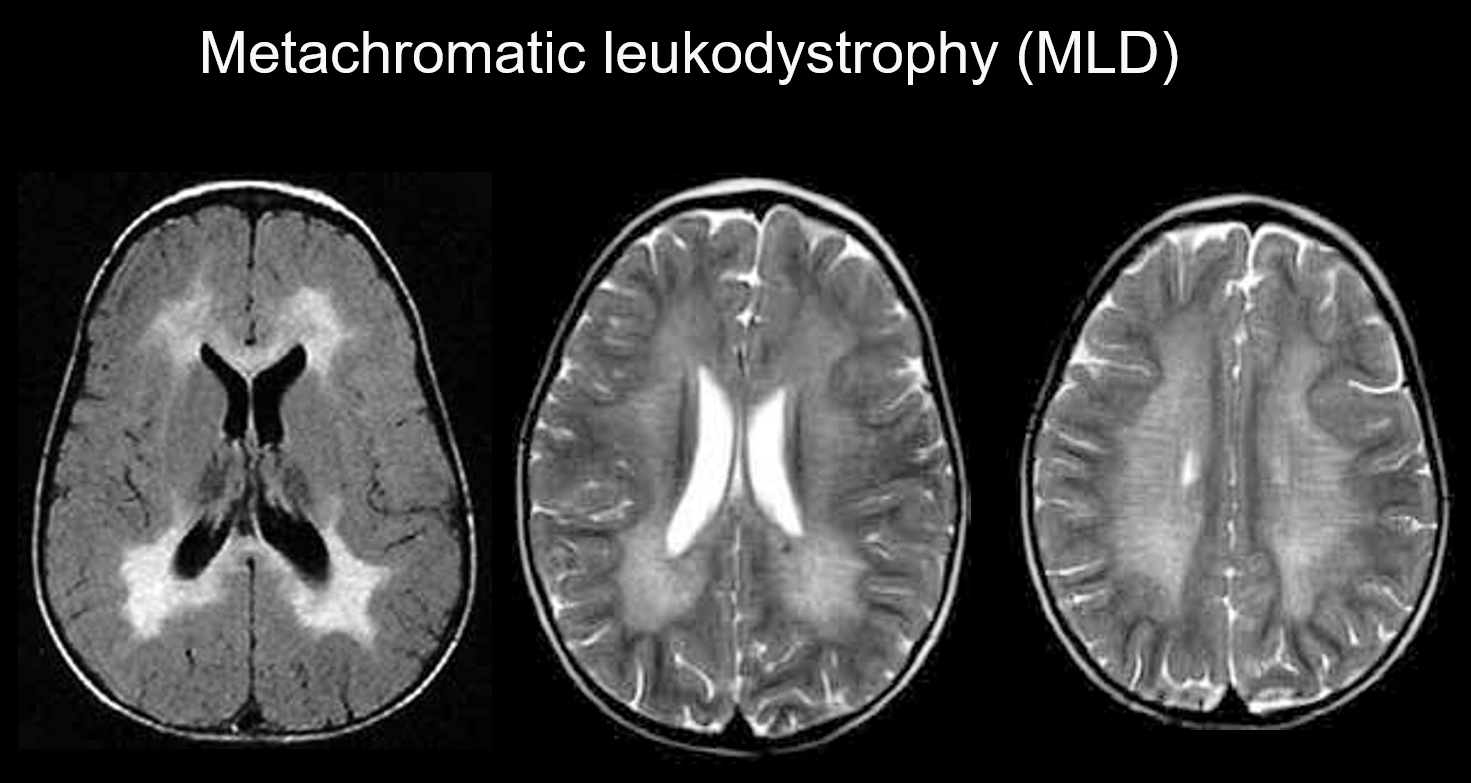
Metachromatická leukodystrofie je autozomálně recesivně dědičné onemocnění (porucha lysozomálního ukládání) způsobené deficitem arylsulfatázy-A (chromozom 22q13.31), při kterém dochází k hromadění vysoce toxického sulfatidu, což má za následek demyelinizaci. V závislosti na věku, ve kterém se objeví, se klasifikuje jako vrozená, s dětským začátkem, s juvenilním začátkem nebo s dospělým začátkem. Mezi její příznaky patří regrese kognitivních funkcí, spastická obrna, mimovolní pohyby, periferní neuropatie a atrofie zrakového nervu. Na T2 váženém zobrazení se projevuje jako hyperintenzita bílé hmoty především v okolí postranních komor a na T1 váženém zobrazení jako mírná hypointenzita. Léze bývají převážně ve frontálním laloku. V rámci rozsáhlých abnormálních signálů v bílé hmotě mohou být patrné pásy normální intenzity (tygří pruhy) (obrázek 5). Předpokládá se, že jsou způsobeny částečným zachováním myelinové pochvy v perivaskulárním prostoru a nahromaděním produktů rozpadu myelinové pochvy v makrofázích.
5. Subkortikální převaha
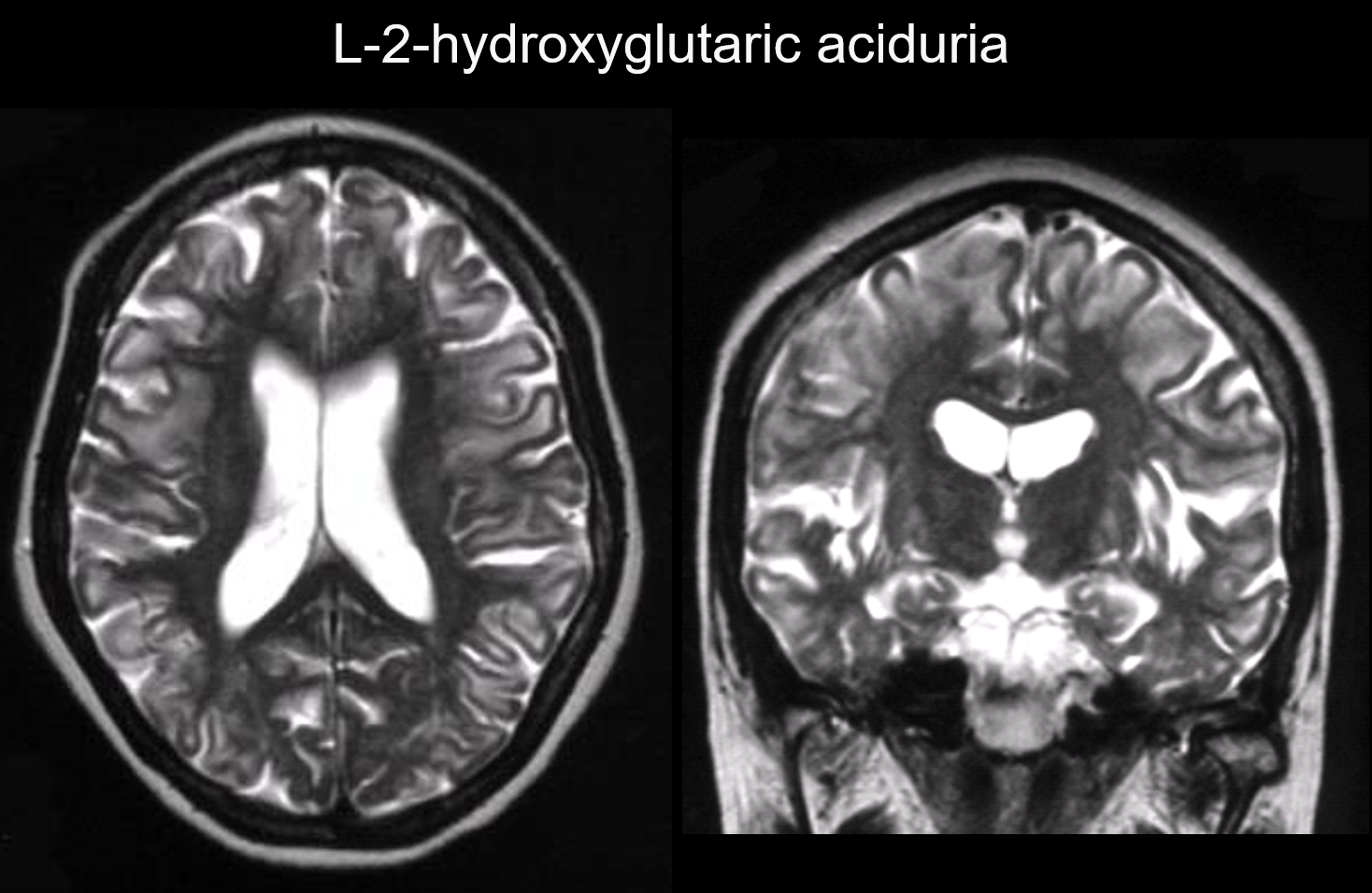
U těchto poruch se léze vyskytují především v subkortikální bílé hmotě, včetně U-vláken. Mezi poruchy s tímto obrazem patří L-2-hydroxyglutarová acidurie (obrázek 6), galaktosemie, Kearnsův-Sayerův syndrom, propionová akademie, poruchy cyklu močoviny a Canavanova choroba v raném stadiu
6. Difuzní mozkové
Při těchto poruchách se abnormální signály objevují v celé bílé hmotě mozku. Vykazují silnou hyperintenzitu T2 ve srovnání se signály T2 produkovanými nemyelinizovanou bílou hmotou (hypomyelinizace). Kromě případů megalencefalické leukoencefalopatie se subkortikálními cystami a leukoencefalopatie s mizející bílou hmotou se u pacientů s jakýmkoli typem poruchy bílé hmoty nakonec s postupujícím onemocněním objeví tento obraz.
(a) Megalencefalická leukoencefalopatie se subkortikálními cystami (MLC)
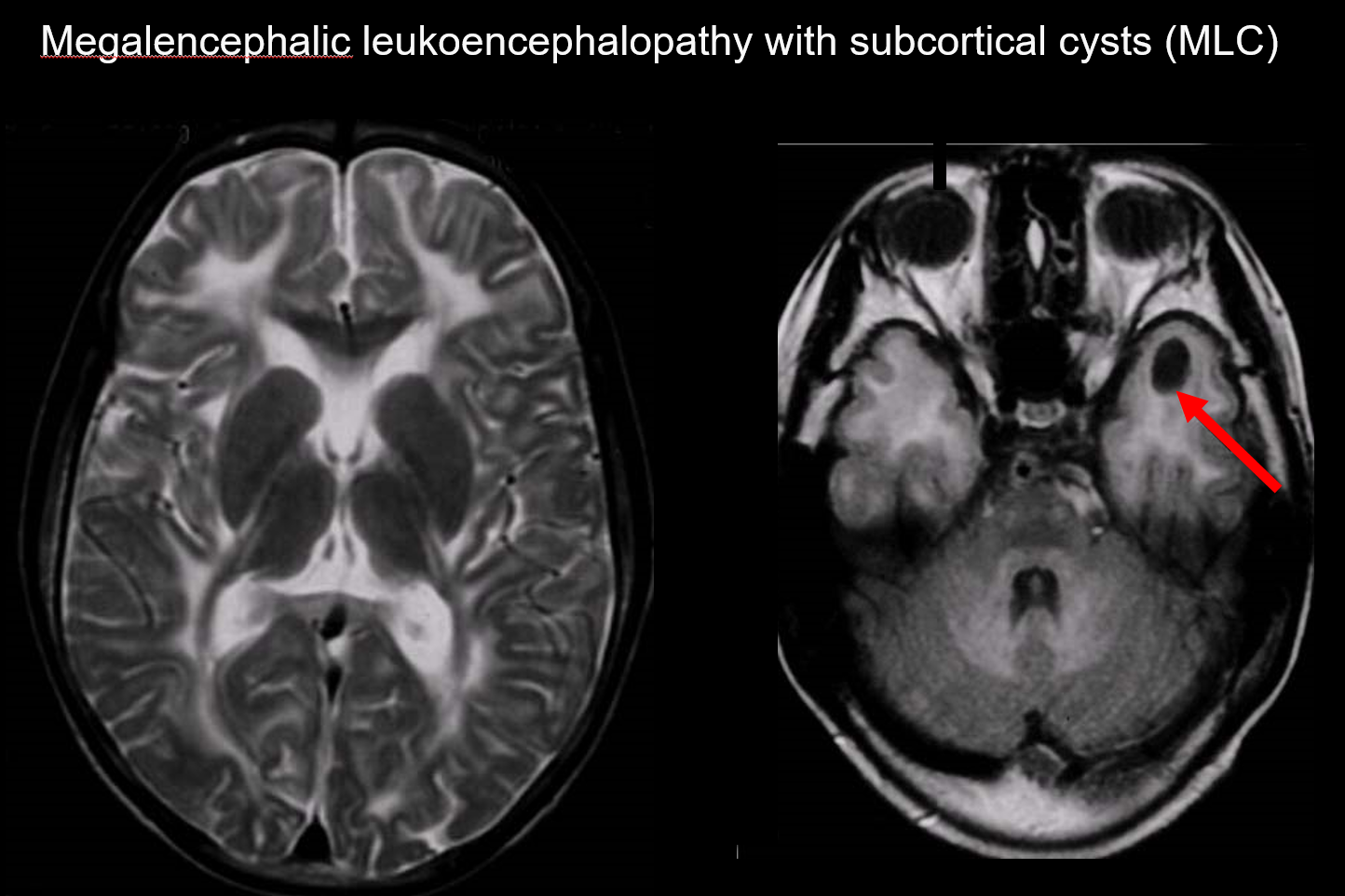
MLC je autozomálně recesivně dědičná porucha způsobená abnormalitou genu MLC1 a její nástup v kojeneckém věku se vyznačuje megalocefalií, pomalu postupující motorickou deteriorací, ataxií a spasticitou. MRI odhaluje charakteristické rozsáhlé abnormální signály v bílé hmotě a mírný otok bílé hmoty, jakož i tvorbu cyst v parietálním a temporálním laloku (obr. 7).7, 8) T1 vážené a T2 vážené zobrazení odhaluje abnormální bílou hmotu, zatímco všechny cysty vykazují T1 hypointenzitu a T2 hyperintenzitu, což je činí obzvláště obtížně detekovatelnými. Pro jeho diagnostiku je cenné zobrazení FLAIR, které zobrazuje cysty (vodu) jako hypointenzity. U Japonců je častější než vakuolizující megalencefalická leukoencefalopatie.
(b) Leukoencefalopatie s mizející bílou hmotou.
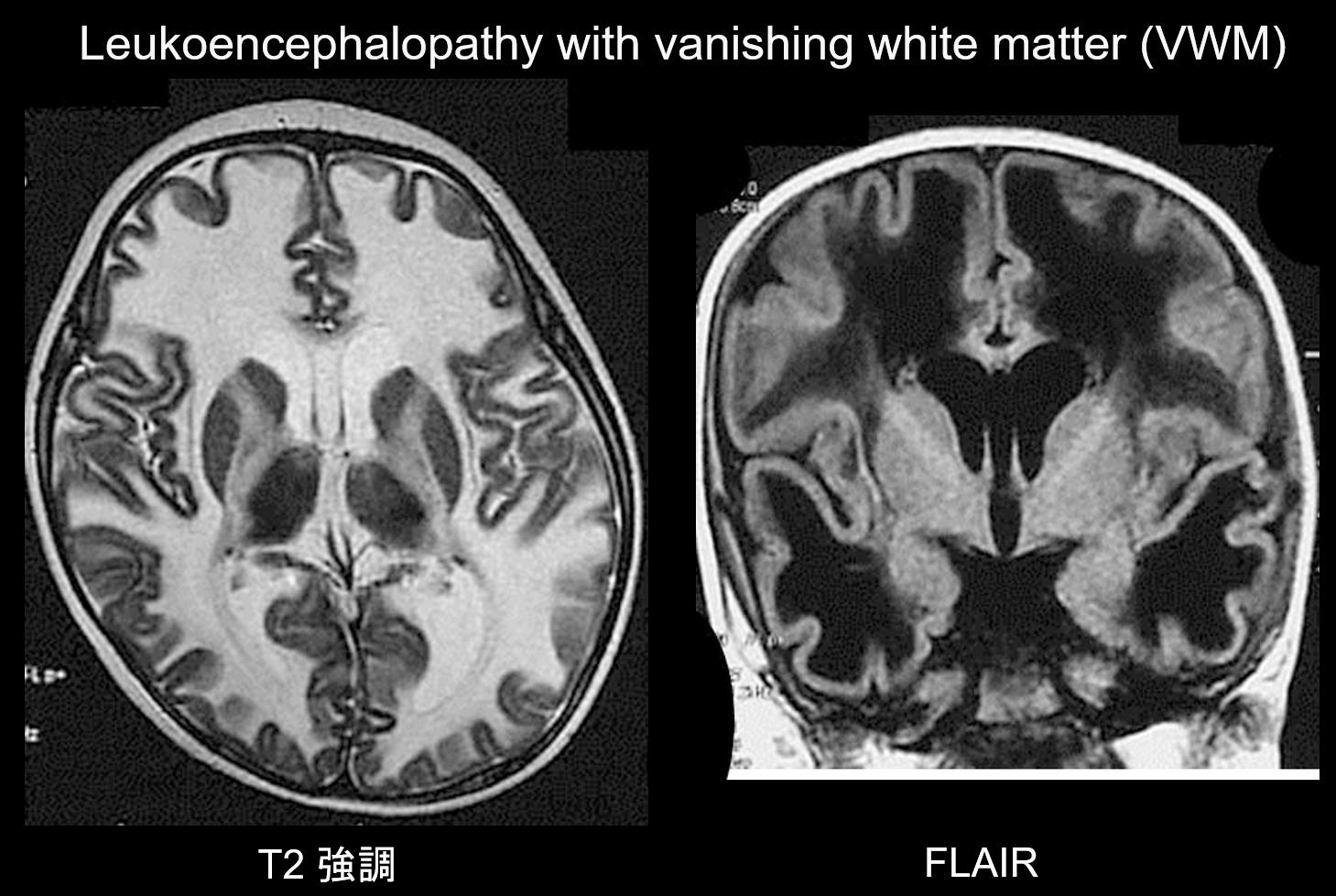
Leukoencefalopatie s mizející bílou hmotou (VWM) je autozomálně recesivně dědičná porucha způsobená nedostatkem eIF2B, proteinu spojeného s eIF2, který přenáší iniciátorovou tRNA na ribozomy. eIF2B se skládá z pěti různých proteinů, které mají různé genetické lokusy. Bylo prokázáno, že VWM je stejná porucha jako dětská mozečková ataxie a centrální hypomyelinizace (CACH). Pacienti jsou v novorozeneckém období a v raném dětství normální, ale po nástupu onemocnění (obvykle ve věku 2-6 let) se u nich rozvíjí pomalu progredující kognitivní regrese, spasticita a ataxie. Je známo, že tyto příznaky se zhoršují při infekci nebo drobném úrazu. Bílá hmota mozková vykazuje rozsáhlou T2 hyperintenzitu a T1 hypointenzitu a v průběhu času je postupně nahrazována tekutinou (jak název napovídá, bílá hmota mizí) (obr. 8). Cystická bílá hmota obsahuje pruhovité struktury, o nichž se předpokládá, že představují zbývající tkáň. Abnormální signály jsou patrné také v mozkovém kmeni, zejména v centrálním tegmentálním traktu. Pro diagnózu této poruchy je cenné zobrazení FLAIR.
7. Převaha nebo prominence zadní jámy
Tyto poruchy jsou charakterizovány lézemi převážně v mozkovém kmeni a mozečku. Léze bílé hmoty mozečku mohou být způsobeny poruchami zahrnujícími cerebrotendinózní xantomatózu (CTX), peroxisomální poruchy, Alexandrovu nemoc, leukoencefalopatii s postižením mozkového kmene a míchy a elevací laktátu (LBSL), onemocnění javorového sirupu v moči, histiocytózu a toxicitu heroinu a kokainu. Léze mozkového kmene mohou být způsobeny poruchami, jako je Alexandrova choroba, LSBL a polyglukosanová choroba dospělých. Léze středního mozečku se vyskytují u syndromu křehkého X a autozomálně dominantní leukodystrofie dospělých související s duplikací lamin B1.
8. Multifokální léze
Na rozdíl od poruch, které způsobují splývající léze popsané v bodech 2-7 výše, poruchy v této části vedou k multifokálním (roztroušeným) lézím. Patří mezi ně infekce, jako je TORCH syndrom (v důsledku vrozené cytomegalovirové infekce nebo jiné příčiny) a brucelóza; zánětlivé poruchy, jako je akutní diseminovaná encefalomyelitida (ADEM), roztroušená skleróza (RS) a neuromyelitis optica (NMO); vaskulopatie, jako je mozková autozomálně dominantní arteriopatie se subkortikálními infarkty a leukoencefalopatií (CADASIL), ateroskleróza, amyloidní angiopatie, mozková choroba malých cév spojená s COL4A1, Fabryho choroba a Susacův syndrom; a dědičná onemocnění, jako je mitochondriální onemocnění, L-2-hydroxyglutarová acidurie, mukopolysacharidóza (MPS) a chromozomální abnormality (např. 6p-syndrom).
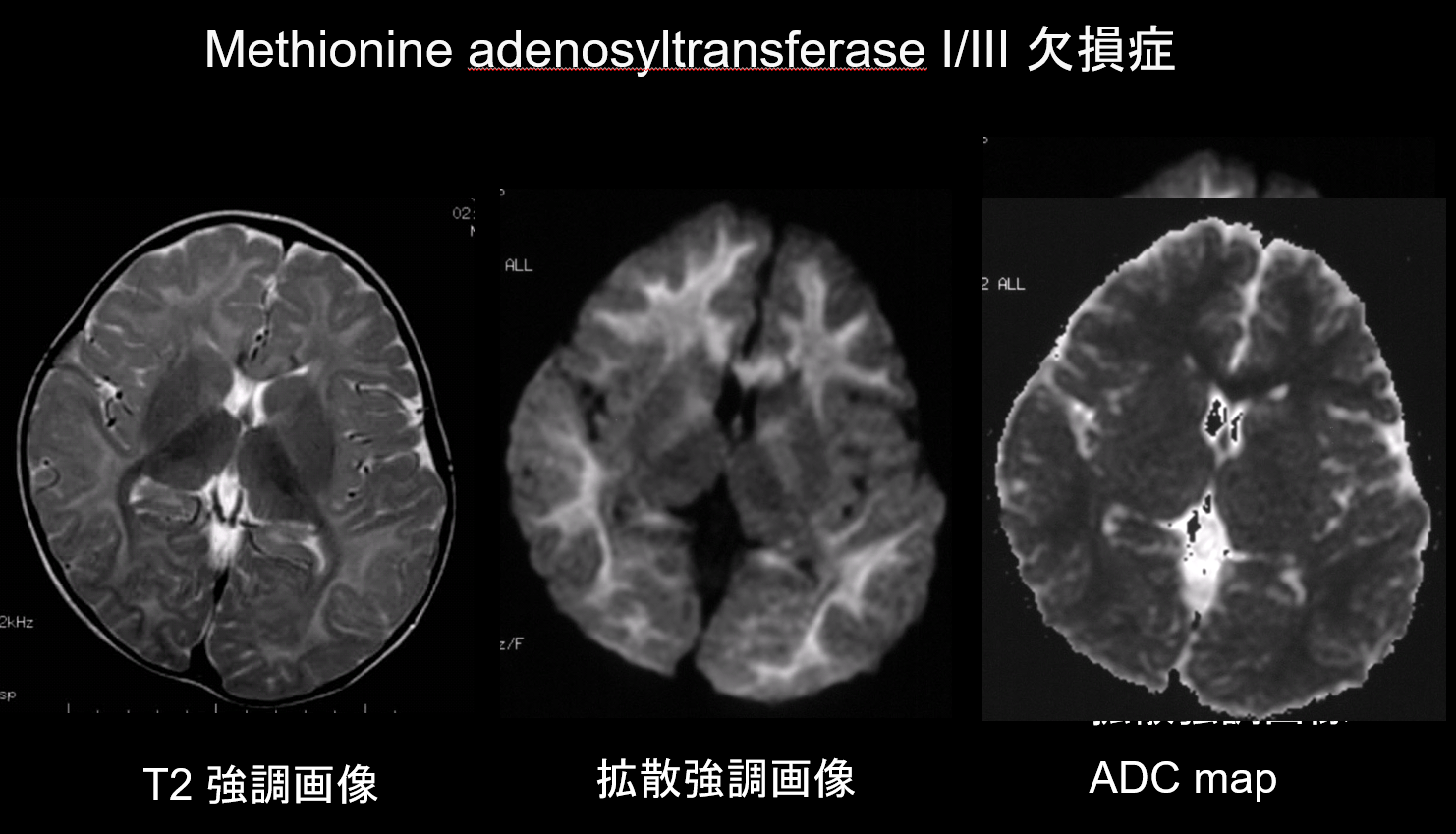
9. Léze s nízkou difuzní kapacitou
Při demyelinizaci i hypomyelinizaci, hlavních patologických stavech poruch bílé hmoty, dochází ke snížení množství myelinu, což omezuje difuzi, a odpovídající zvýšení množství extracelulární tekutiny má za následek T2 hyperintenzity s vysokým zdánlivým difuzním koeficientem (ADC). U poruch bílé hmoty se vzácně vyskytují jak hyperintenzity T2, tak nízký ADC, a proto má tato kombinace vysokou diagnostickou hodnotu. Poruchy charakterizované přítomností intramyelinického edému v myelinových pochvách a v mezerách mezi pochvami vykazují nízký ADC. Patří mezi ně nemoc z javorového sirupu, nedostatek methionin adenosyltransferázy I/III (obrázek 9), fenylketonurie, neketotická hyperglycinemie a Canavanova nemoc. Krabbeho choroba a metachromatická leukodystrofie mohou také vykazovat nízkou ADC v některých lézích bílé hmoty, protože v akutní fázi demyelinizace může dojít k intramyelinickému edému.
- Van der Knaap MS, Valk J. Classification of myelin disorders. In Van der Knaap MS, Valk J, eds. Magnetická rezonance myelinizace a poruchy myelinu. Berlin. 3. vyd: Springer, 2005, 20-24.
- Schiffmann R, van der Knaap MS. Přístup k diagnostice poruch bílé hmoty založený na magnetické rezonanci. Neurology 2009; 72: 750-759
- Takanashi J. Diagnostické zobrazování poruch bílé hmoty. Journal of the Japan Pediatric Society 2007; 111: 1243-1254.
- Van der Knaap MS, Breiter SN, Naidu S, et al. Definování a kategorizace leukoencefalopatií neznámého původu: Přístup k zobrazování pomocí magnetické rezonance. Radiology 1999; 213: 121-133.
.