Diagnosticarea imagistică a anomaliilor materiei albe | Hipomielinizarea cerebrală congenitală; rețea pentru boala Pelizaeus-Merzbacher și afecțiuni înrudite
Diagnostic Imaging of White Matter Abnormalities
Junichi TAKANASHI, Departamentul de pediatrie, Tokyo Women’s Medical University, Yachiyo Medical Center
Introducere
În această lucrare, prezint abordarea utilizată pentru diagnosticarea tulburărilor care apar ca semnale anormale în materia albă cerebrală la imagistica prin rezonanță magnetică (IRM), de la imagistică la diagnostic. Tulburările care afectează în principal substanța albă sunt denumite în general „leucoencefalopatie” sau „white matter disorders” în limba engleză1, 2) 3) Un alt termen, leucodistrofie, este uneori confundat cu degenerescența materiei albe, dar acesta se referă de fapt la un spectru mai restrâns de afecțiuni cu o componentă genetică (afecțiuni demielinizante moștenite).
Clasificarea bazată pe imagistică a afecțiunilor materiei albe
Avântul IRM a îmbunătățit în mod dramatic capacitatea noastră de a detecta leziunile din materia albă a sistemului nervos central. Multe forme cunoscute de tulburări ale substanței albe prezintă semne specifice la IRM, ceea ce este util pentru diagnosticarea lor. Identificarea modelelor de anomalii ale substanței albe observate la IRM (imagistică cu ponderare T1, cu ponderare T2 sau FLAIR) facilitează restrângerea posibilităților în multiplele diagnostice diferențiale. Clasificarea lui Schiffmann și van der Kamp a tulburărilor de substanță albă în funcție de constatările IRM are o valoare practică2, 4) (Figura 1, Tabelul 1). Chiar dacă aceasta nu conduce la un diagnostic final, clasificarea constatărilor imagistice poate duce la descoperirea ulterioară a unei noi afecțiuni. În cele ce urmează, descriu tulburările de substanță albă în termenii clasificărilor IRM de mai sus și explic principalele tipuri de tulburări.
Tabel 1. Lista tulburărilor în funcție de tiparele IRM
- Predominanță frontală
Boala Alexander, varianta frontală a adrenoleucodistrofiei legate de X (ALD), leucodistrofia metacromatică (MLD), leucodistrofia neuroaxonală cu sferoizi. - Predominanță parieto-occipitală
Adrenoleucodistrofie legată de X (ALD), boala Krabbe,tulburări peroxisomale cu debut precoce, hipoglicemie neonatală. - Predominanță periventriculară
Leucodistrofia metacromatică (MLD), boala Krabbe, sindromul Sjögren-Larsson, boala cu corpuri poliglucoziene la adult, leucoencefalopatie cu afectarea trunchiului cerebral și a măduvei spinării și creșterea lactatelor (LBSL), leucomalacie periventriculară (PVL), encefalopatie HIV, lipofuscinoze ceroide neuronale cu debut tardiv. - Predominanță subcorticală
Acidurie L-2-hidroxiglutariană, galactosemie, sindromul Kearns-Sayer, academia propionică, tulburări ale ciclului ureei, boala Canavan. - Leucoencefalopatie cerebrală difuză
Leucoencefalopatie megalencefalică cu chisturi subcorticale (MLC), leucoencefalopatie cu dispariție a substanței albe (VWM), distrofie musculară congenitală cu deficit de merosină, boală mitocondrială, deficit de cofactor molibden, deficit de sulfitoxidază, cazuri avansate de tulburări ale substanței albe. - Predominanța sau proeminența fosei posterioare
Lesiuni ale cerebelului și ale pedunculilor cerebeloși: xantomatoza cerebrotendinoasă (CTX), tulburări peroxisomale, boala Alexander, leucoencefalopatie cu afectare a trunchiului cerebral și a măduvei spinării și creștere a lactatelor (LBSL), boala urinară cu sirop de arțar, histiocitoza, leucodistrofia autozomal dominantă a adultului legată de o duplicare a laminului B1, toxicitate la heroină și cocaină.
Lesiuni ale trunchiului cerebral: Boala Alexander, LSBL, tulburări peroxisomale, boala Wilson, boala poliglucozică a adultului, sindromul Leigh, atrofia dentatorubropallidoluysiană (DRPLA), boala corpului poliglucozică a adultului, leucodistrofia autozomal dominantă a adultului legată de o duplicare a laminelor B1. - Leziuni multifocale
SindromulTORCH (infecție congenitală cu citomegalovirus), bruceloză, encefalomielită acută diseminată (ADEM), scleroză multiplă (SM), neuromielită optică (NMO), arteriopatie cerebrală autosomal-dominantă cu infarcte subcorticale și leucoencefalopatie (CADASIL), ateroscleroză, angiopatie amiloidă, boală cerebrală a vaselor mici asociată cu COL4A1, boala Fabry, sindromul Susac, boli mitocondriale, acidurie L-2-hidroxiglutariană, mucopolizaharidoză (MPS), anomalii cromozomiale (cum ar fi sindromul 6p). - Leziuni cu capacitate de difuzie scăzută
Boala de urină cu sirop de arțar, deficitul de metionină adenoziltransferază I/III, fenilcetonuria, hiperglicinemia necetotică, boala Canavan, leziuni active în boala Krabbe și leucodistrofia metacromatică.
1. Hipomielinizarea materiei albe cerebrale
Se referă la un grup de afecțiuni în care formarea tecii de mielină este afectată sau întârziată, iar imaginile sale seamănă cu cele ale nou-născuților cu mielinizare imatură. Pe imaginile ponderate în T2, substanța albă apare în mod caracteristic ca o hiperintensitate răspândită care este slabă în comparație cu cortexul. Pentru mai multe detalii, consultați site-ul web al hipomielinizării cerebrale congenitale.
Dacă leziunile de substanță albă nu sunt compatibile cu hipomielinizarea materiei albe cerebrale, trebuie stabilit dacă sunt confluente sau multiple.2) Leziunile confluente de substanță albă se datorează, de obicei, degenerării moștenite a substanței albe (leucodistrofie) și, în majoritatea cazurilor, sunt simetrice bilateral. Leziunile multiple ale substanței albe sunt de obicei asimetrice și dobândite. Leziunile confluente ale substanței albe sunt în continuare subdivizate în categoriile 2-7 de mai jos.
2. Predominanța frontală
În acest grup de afecțiuni, leziunile extinse ale substanței albe sunt prezente predominant în lobul frontal. Acestea includ boala Alexander, varianta frontală a adrenoleucodistrofiei (ALD) legată de X, leucodistrofia metacromatică (MLD) și leucodistrofia neuroaxonală cu sferoizi.
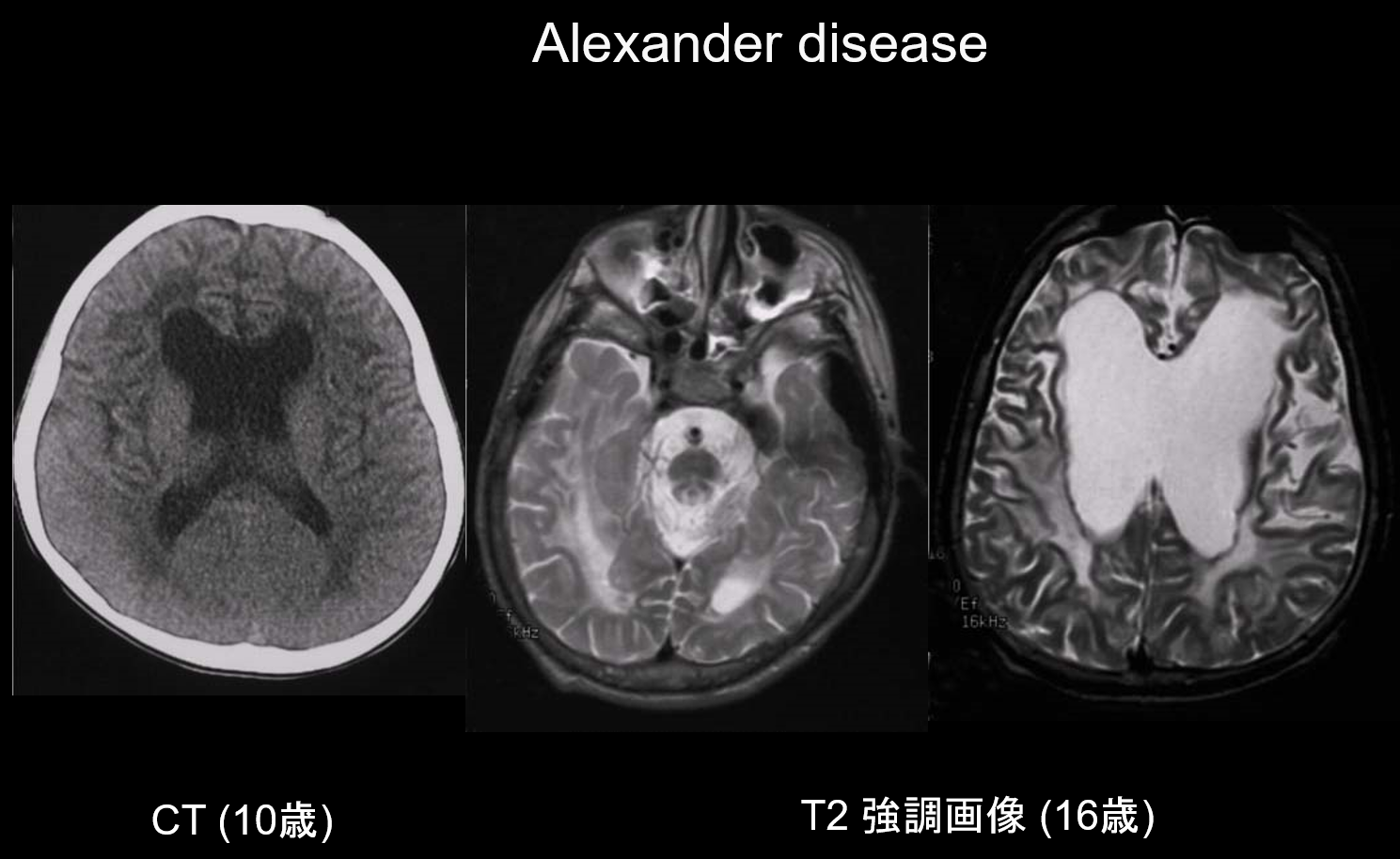
(a) Boala Alexander.
Boala Alexander este o tulburare ereditară autozomal dominantă cauzată de o mutație în gena GFAP de pe cromozomul 17q21. Ea are ca rezultat acumularea de fibre Rosenthal în celulele gliale stelate. Aceste fibre sunt compuse din GFAP și proteine de stres (αB-cristalin și HSP27. Boala Alexander apare în principal în copilărie, între vârstele de 3 luni și 2 ani, cu apariția megalencefaliei, a retardului de dezvoltare, a paraliziei spastice și a epilepsiei. La IRM, poate prezenta (i) leziuni extinse ale substanței albe, predominant în lobul frontal; (ii) marginalizare T1 hiperintensă și T2 hipointensă în jurul ventriculilor laterali; (iii) leziuni în ganglionii bazali și talamus; (iv) leziuni ale trunchiului cerebral; și (v) intensificarea prin contrast a leziunilor active (figura 2). În stadiile incipiente, alături de leziuni ale substanței albe și ale putamenului, se observă umflături care pot cauza treptat atrofie sau formarea de chisturi.
3. Predominanța parieto-occipitală
Caracteristica principală a acestui grup de tulburări este reprezentată de leziuni ale substanței albe parieto-occipitale. Ele includ adrenoleucodistrofia legată de X (ALD), boala Krabbe, tulburările peroxisomale cu debut precoce și hipoglicemia neonatală.
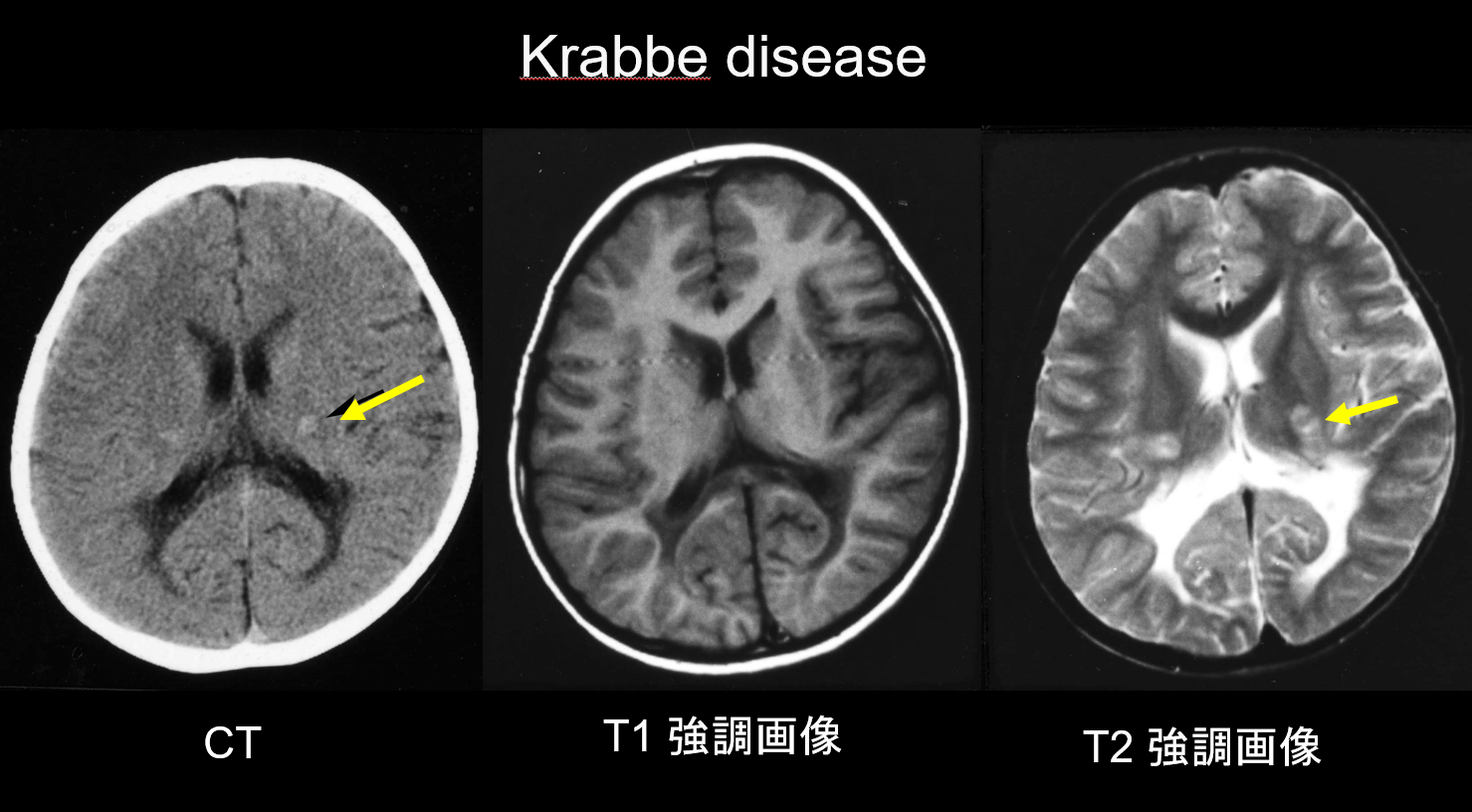
(a) Boala Krabbe.
Boala Krabbe este o afecțiune ereditară autozomal recesivă (boală de depozitare lizozomală) cauzată de deficitul de galactosilceramidază (cromozomul 14q31), în care se crede că acumularea de psihozină foarte citotoxică provoacă demielinizarea generalizată. Apar, de asemenea, celule mari, multinucleate, numite „celule globoide”. În funcție de vârsta la care apare, este clasificată ca boală infantilă, infantilă cu debut tardiv, juvenilă sau cu debut la adult. Cele mai multe cazuri sunt infantile și debutează cu apariția febrei, iritabilitate, dificultăți de hrănire, retard de dezvoltare, neuropatie periferică, spasticitate și atrofie a nervului optic la vârsta de 3-6 luni. În stadiile incipiente, tomografia computerizată (CT) relevă o hiperdensitate caracteristică în talamus și corona radiata. Se crede că aceasta reflectă celulele globoide de înaltă densitate și proliferarea glială. IRM poate arăta, de asemenea, hiperintensitate T1 și hipointensitate T2 în jurul ventriculilor, precum și structuri liniare similare cu cele observate în MLD (figura 3). Nucleul dentat cerebelos, substanța albă cerebeloasă și tractul piramidal al trunchiului cerebral prezintă hiperintensitate T2 încă dintr-un stadiu incipient.
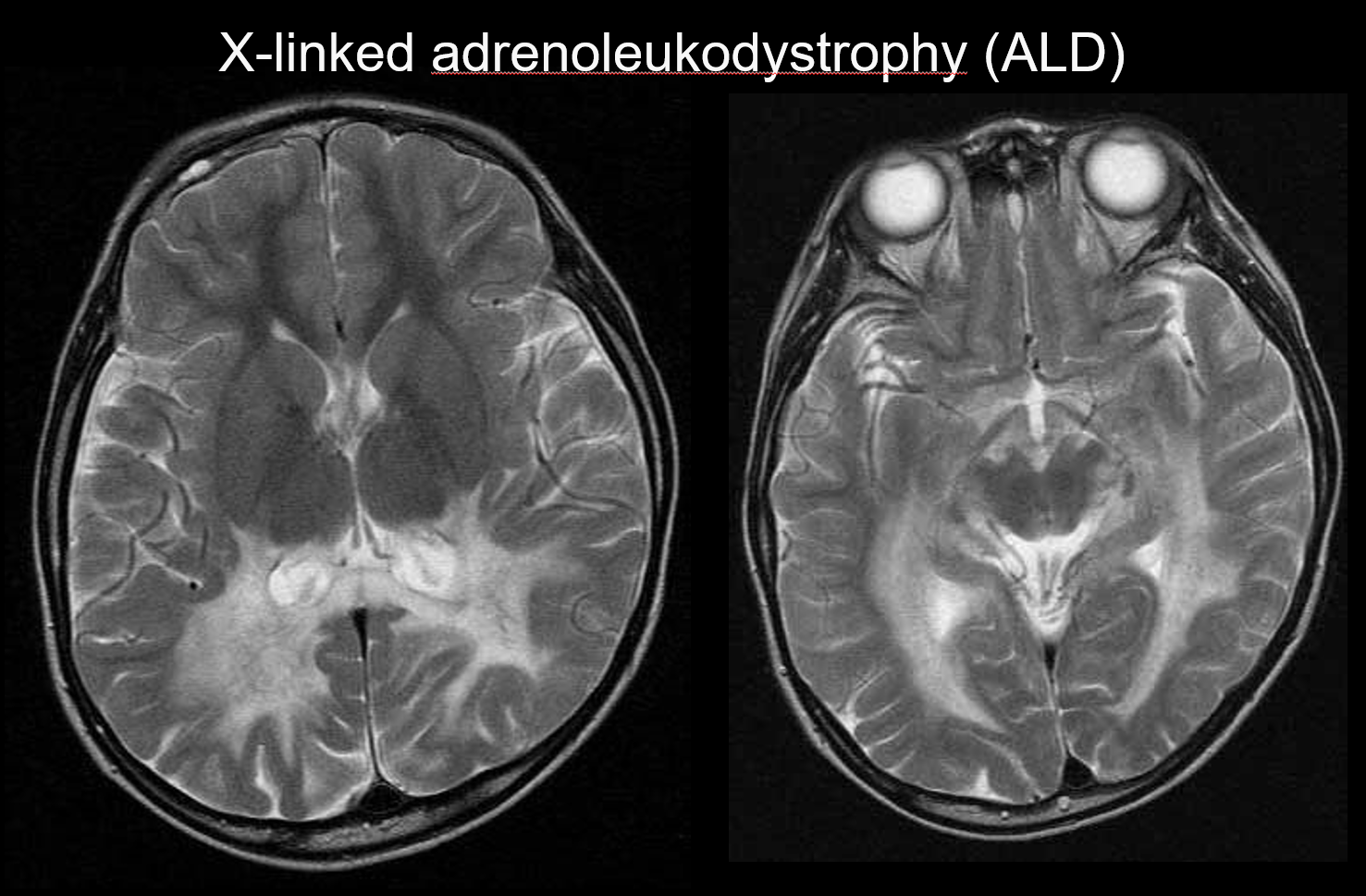
(b) Adrenoleucodistrofia legată de X
Adrenoleucodistrofia legată de X (ALD) este o tulburare ereditară recesivă legată de X (tulburare peroxisomală) cauzată de o anomalie a genei ABCD1 (cromozomul Xq28). Deficiența β-oxidării duce la acumularea de acizi grași cu lanț foarte lung în substanța albă cerebrală și în glandele suprarenale, provocând demielinizare și insuficiență suprarenală. ALD este clasificată în forme cerebrale pentru copii, adolescenți și adulți, adrenomieloneuropatie (AMN) și numai boala Addison. Forma cerebrală a copilăriei se dezvoltă la vârsta de 5-8 ani, cu apariția unor simptome care includ deteriorarea intelectuală, mers spastic și afectarea vederii și auzului. Din punct de vedere patologic, demielinizarea progresează din substanța albă care înconjoară trigonul ventriculului lateral până la spleniul corpului calos, extinzându-se treptat în sens antero-lateral. Reflectând patologia bolii, la IRM sunt vizibile hiperintensități T2 simetrice și hipointensități T1 simetrice care se extind anterolateral din substanța albă care înconjoară trigonul ventriculului lateral, cu intensificare de contrast evidentă la margini (figura 4). Leziunile tractului corticospinal sunt, de asemenea, evidente.
4. Predominanța periventriculară
Aceste afecțiuni se caracterizează în principal prin leziuni în substanța albă care înconjoară ventriculii laterali, substanța albă subcorticală (fibrele U) fiind conservată. Acest model este întâlnit în numeroase tulburări, inclusiv în MLD, și este, prin urmare, relativ nespecific. Semnale ușor anormale în jurul ventriculilor laterali sunt, de asemenea, observate în degenerescența corticală, în special în lipofuscinozele ceroide neuronale care se dezvoltă după copilărie.
(a) Leucodistrofia metacromatică.
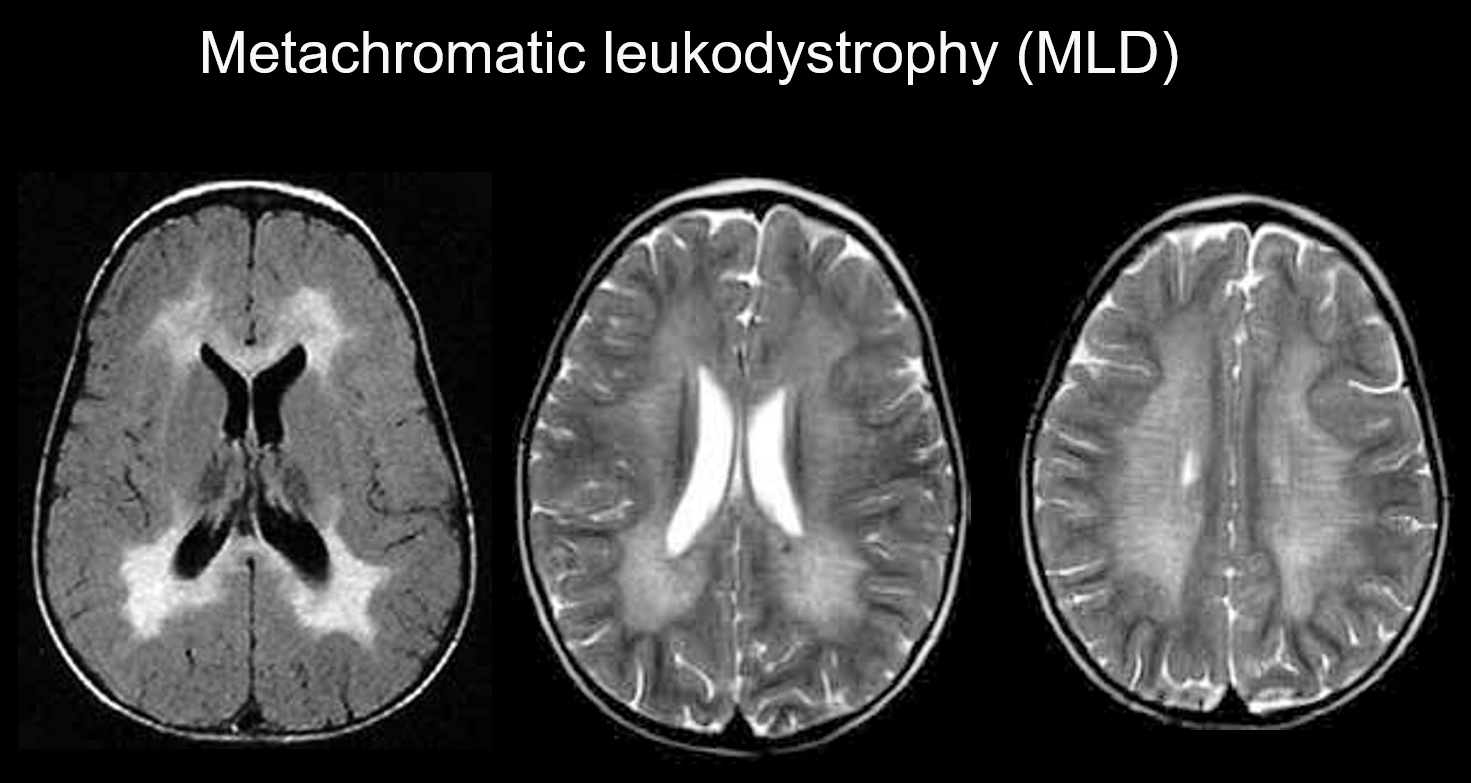
Lucodistrofia metacromatică este o afecțiune ereditară autozomal recesivă (tulburare de depozitare lizozomală) cauzată de deficitul de arilsulfatază-A (cromozomul 22q13.31), în care acumularea de sulfatidă foarte toxică duce la demielinizare. În funcție de vârsta la care apare, este clasificată ca fiind congenitală, cu debut infantil, cu debut juvenil sau cu debut la vârsta adultă. Simptomele sale includ regresie cognitivă, paralizie spastică, mișcări involuntare, neuropatie periferică și atrofie a nervului optic. Apare pe imagistica ponderată în T2 ca hiperintensități ale substanței albe, în principal în jurul ventriculilor laterali, iar pe imagistica ponderată în T1 ca hipointensități ușoare. Leziunile tind să fie predominant în lobul frontal. Benzile de intensitate normală (dungi de tigru) pot fi evidente în cadrul semnalelor anormale răspândite în substanța albă (figura 5). Se crede că acestea se datorează conservării parțiale a tecii de mielină în spațiul perivascular și acumulării de produși de degradare a tecii de mielină în macrofage.
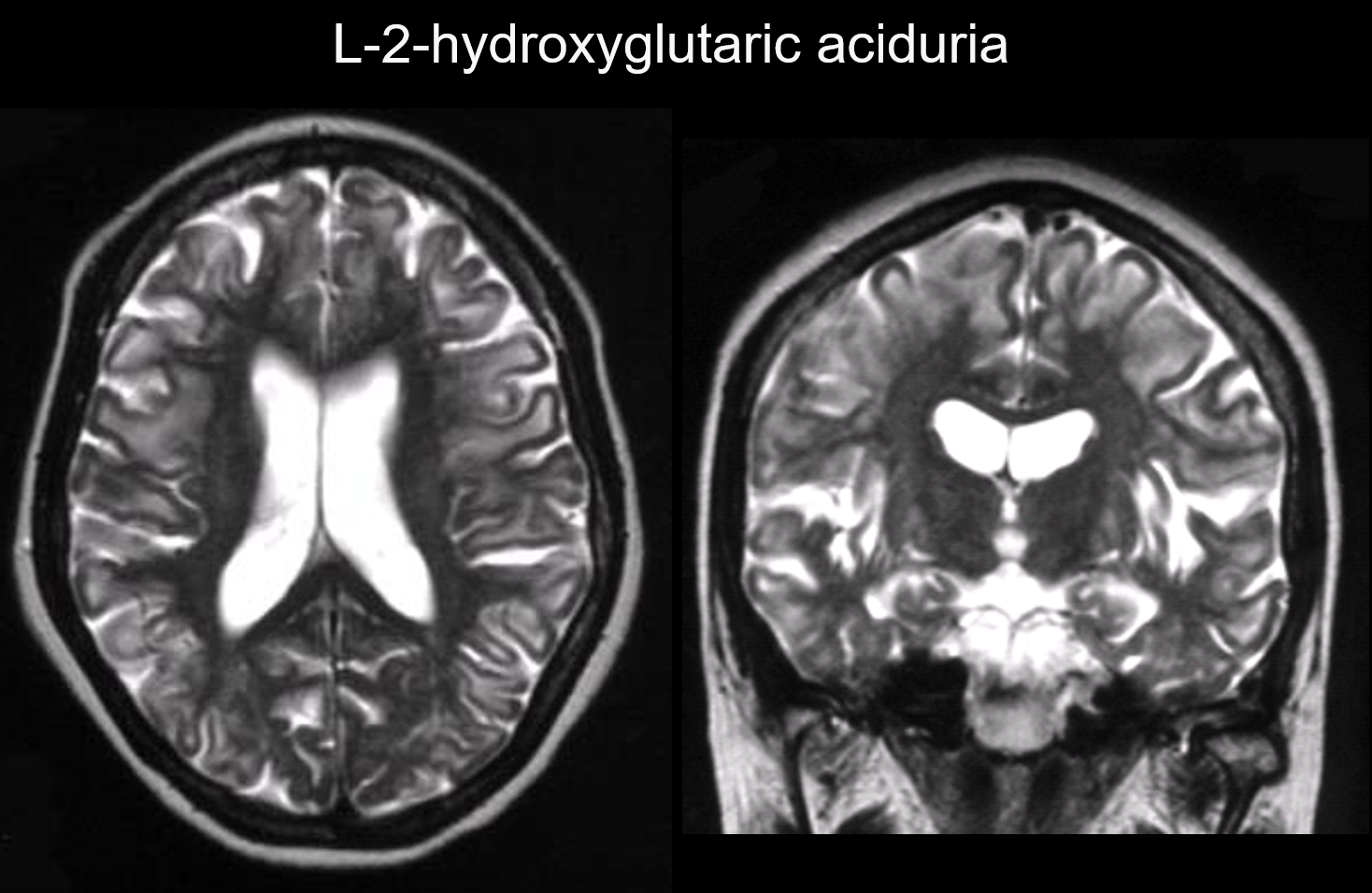
5. Predominanța subcorticală
În aceste tulburări, leziunile apar în principal în substanța albă subcorticală, inclusiv în fibrele U. Tulburările cu acest tipar includ aciduria L-2-hidroxiglutariană (figura 6), galactozemia, sindromul Kearns-Sayer, academia propionică, tulburările ciclului ureei și boala Canavan în stadiu incipient.
6. Difuzia cerebrală
În aceste afecțiuni, semnalele anormale apar în toată substanța albă cerebrală. Ele prezintă hiperintensități T2 puternice în comparație cu semnalele T2 produse de substanța albă nemielinizată (hipomielinizare). În plus față de cazurile de leucoencefalopatie megalencefalică cu chisturi subcorticale și leucoencefalopatie cu dispariție a substanței albe, pacienții cu orice tip de afecțiune a substanței albe prezintă în cele din urmă acest tipar pe măsură ce boala avansează.
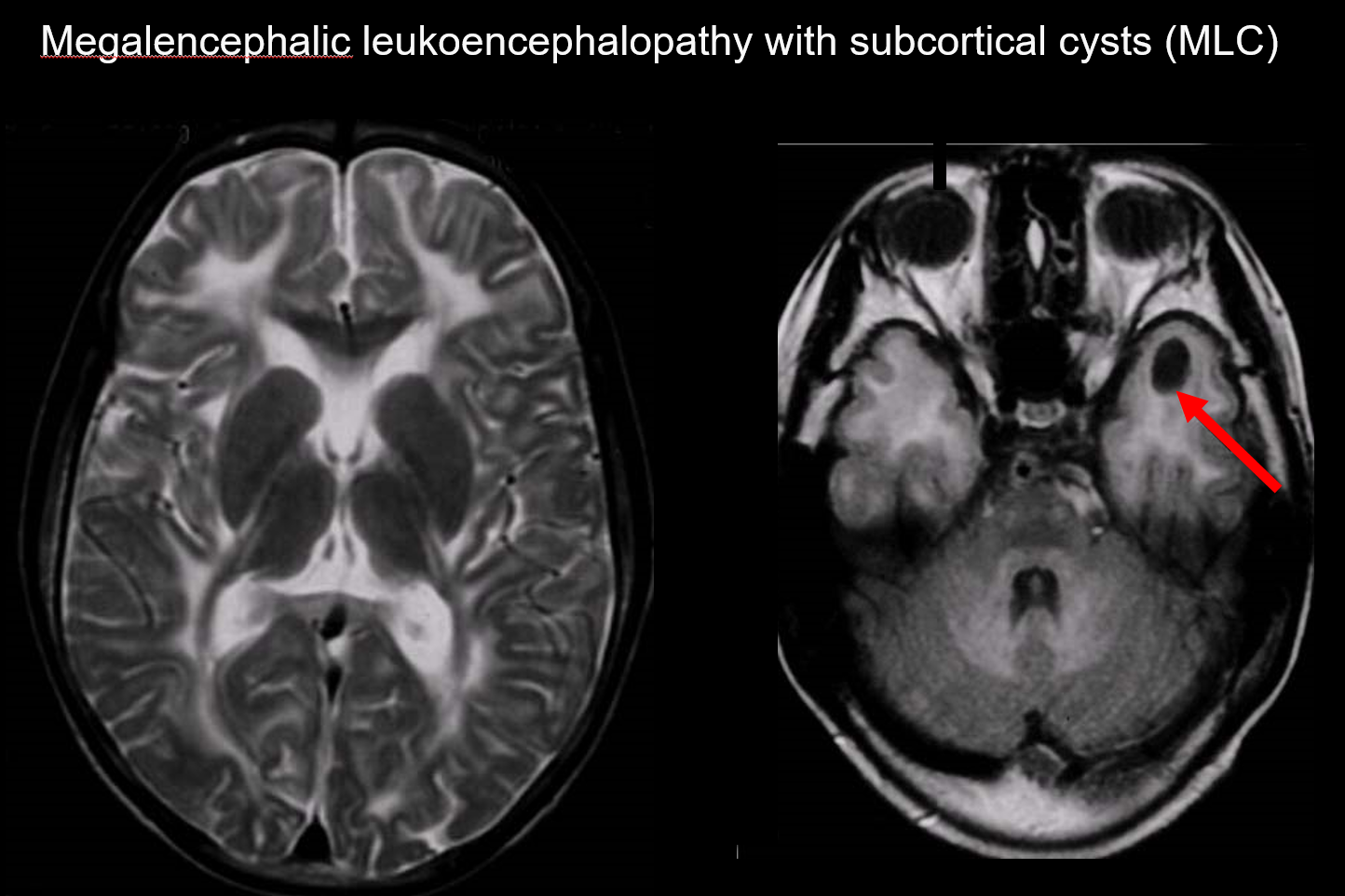
(a) Leucoencefalopatia megalencefalică cu chisturi subcorticale (MLC)
MLC este o tulburare ereditară autozomal recesivă cauzată de o anomalie a genei MLC1, iar debutul său în copilărie este marcat de megalocefalie, deteriorare motorie cu progresie lentă, ataxie și spasticitate. RMN-ul relevă semnale anormale răspândite caracteristice în substanța albă și o ușoară tumefacție a substanței albe, precum și formarea de chisturi în lobii parietali și temporali (figura 7).7, 8) Imagistica ponderată în T1 și în T2 relevă substanța albă anormală, în timp ce chisturile prezintă toate hipointensitate T1 și hiperintensitate T2, ceea ce le face deosebit de dificil de detectat. Imagistica FLAIR, care vizualizează chisturile (apă) ca hipointensități, este valoroasă pentru diagnosticul acesteia. Este mai frecventă în rândul japonezilor decât leucoencefalopatia megalencefalică vacuolantă.
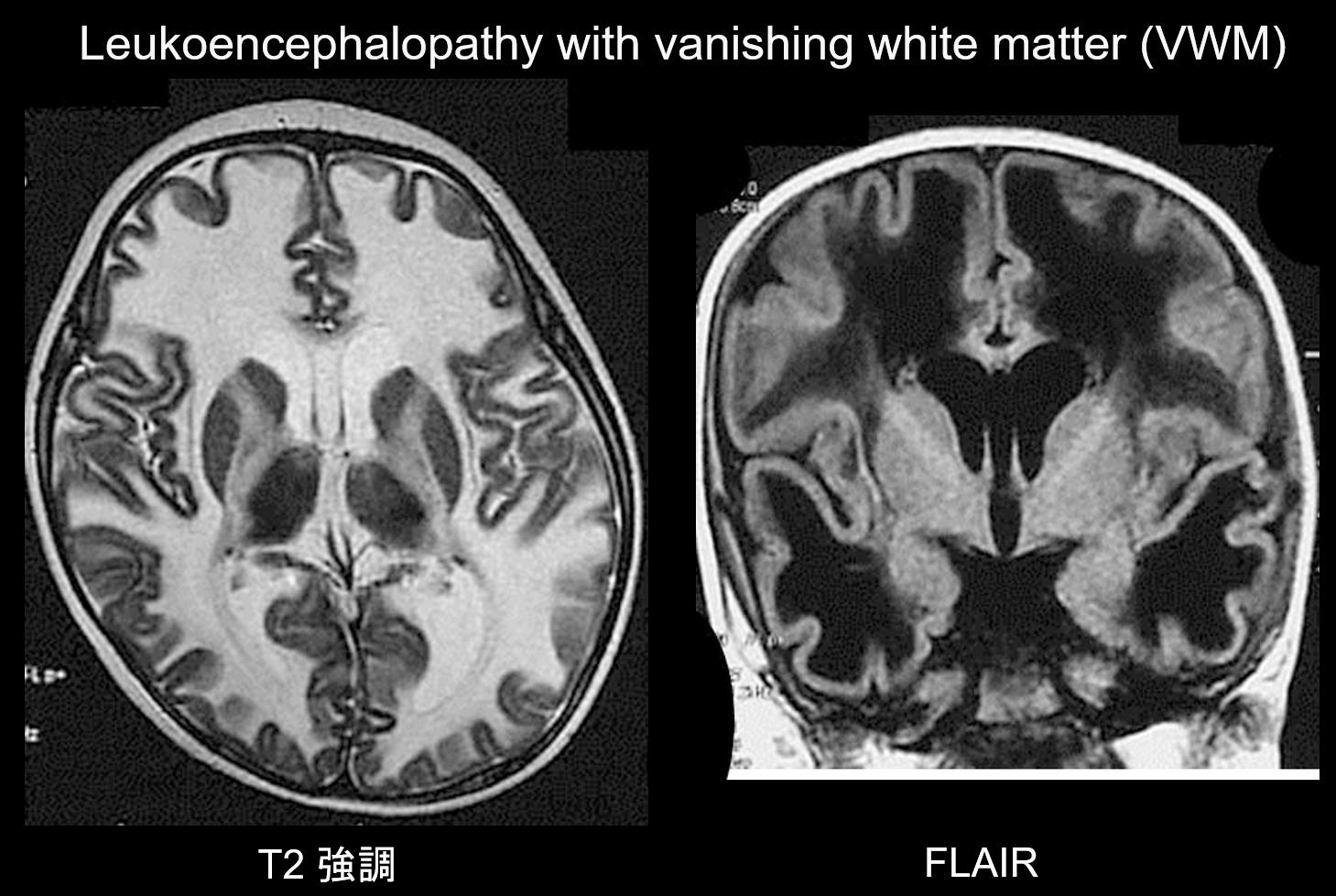
(b) Leucoencefalopatie cu dispariție a substanței albe.
Leukoencefalopatia cu substanță albă evanescentă (VWM) este o tulburare ereditară autosomal recesivă cauzată de un deficit de eIF2B, o proteină asociată cu eIF2, care transferă ARNt inițiator către ribozomi. eIF2B constă în cinci proteine diferite, care au toate loci genetici diferiți. S-a demonstrat că VWM este aceeași tulburare ca și ataxia cerebelară infantilă și hipomielinizarea centrală (CACH). Pacienții sunt normali în perioada neonatală și la începutul copilăriei, dar după debut (de obicei la vârsta de 2-6 ani) dezvoltă regresie cognitivă lent progresivă, spasticitate și ataxie. Se știe că aceste simptome sunt exacerbate de infecții sau traumatisme minore. Substanța albă cerebrală prezintă o hiperintensitate T2 generalizată și o hipointensitate T1 și este înlocuită treptat de lichid în timp (după cum sugerează și numele, substanța albă dispare) (figura 8) (figura 8). Materia albă chistică conține structuri în formă de benzi care se crede că reprezintă țesutul rămas. Semnale anormale sunt, de asemenea, observate în trunchiul cerebral, în special în tractul tegmental central. Imagistica FLAIR este valoroasă pentru diagnosticul acestei tulburări.
7. Predominanța sau proeminența fosei posterioare
Aceste tulburări se caracterizează prin leziuni predominant în trunchiul cerebral și cerebel. Leziunile materiei albe cerebeloase pot fi cauzate de afecțiuni care includ xantomatoza cerebrotendinoasă (CTX), afecțiuni peroxisomale, boala Alexander, leucoencefalopatie cu afectare a trunchiului cerebral și a măduvei spinării și creștere a lactatelor (LBSL), boala siropului de arțar în urină, histiocitoza și toxicitatea heroinei și cocainei. Leziunile trunchiului cerebral pot fi cauzate de tulburări cum ar fi boala Alexander, LSBL și boala poliglucozică a adultului. Leziunile pedunculului cerebelos mijlociu sunt observate în sindromul X fragil și în leucodistrofia autozomal dominantă a adultului legată de o duplicare a laminelor B1.
8. Leziuni multifocale
În comparație cu tulburările care produc leziuni confluente descrise la 2-7 de mai sus, tulburările din această secțiune determină leziuni multifocale (împrăștiate). Acestea includ infecții precum sindromul TORCH (datorat infecției congenitale cu citomegalovirus sau alte cauze) și bruceloza; tulburări inflamatorii precum encefalomielita acută diseminată (ADEM), scleroza multiplă (SM) și neuromielita optică (NMO); vasculopatii, cum ar fi arteriopatia cerebrală autosomal-dominantă cu infarcte subcorticale și leucoencefalopatie (CADASIL), ateroscleroza, angiopatia amiloidă, boala cerebrală a vaselor mici asociată cu COL4A1, boala Fabry și sindromul Susac; și afecțiuni moștenite, cum ar fi boala mitocondrială, aciduria L-2-hidroxiglutariană, mucopolizaharidoza (MPS) și anomalii cromozomiale (cum ar fi sindromul 6p).
9. Leziuni cu capacitate scăzută de difuzie
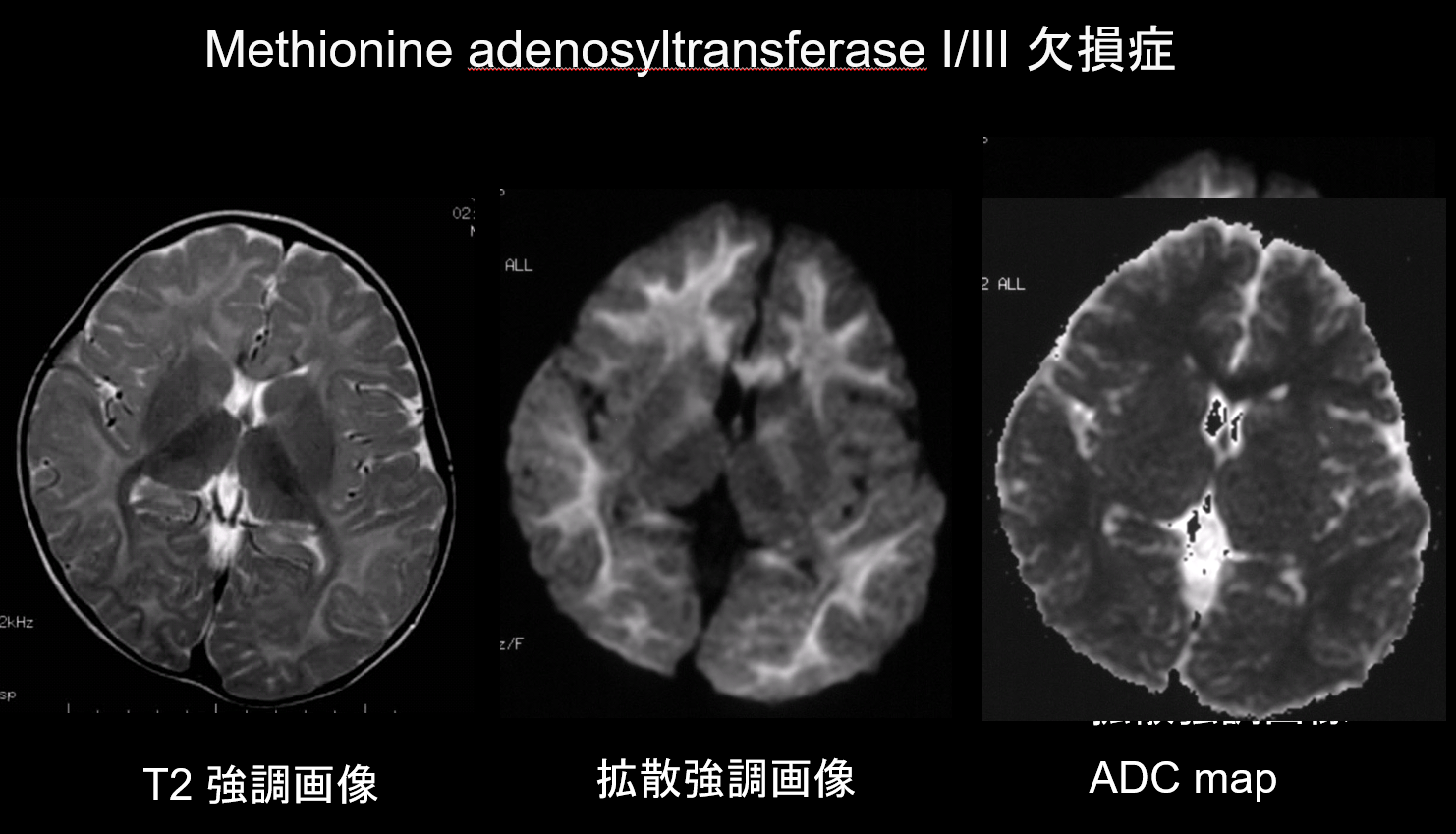
Atât în cazul demielinizării, cât și al hipomielinizării, principalele patologii ale afecțiunilor substanței albe, există o scădere a cantității de mielină, care limitează difuzia, iar creșterea corespunzătoare a fluidului extracelular are ca rezultat hiperintensități T2 cu un coeficient de difuzie aparentă (ADC) ridicat. Este rar ca tulburările de substanță albă să prezinte atât hiperintensități T2, cât și ADC scăzut, iar această combinație are, prin urmare, o valoare diagnostică ridicată. Tulburările caracterizate de prezența edemului intramielinic în interiorul tecii de mielină și în spațiile dintre teci prezintă un ADC scăzut. Acestea includ boala siropului de arțar în urină, deficitul de metionină adenoziltransferază I/III (figura 9), fenilcetonuria, hiperglicinemia non-cetotică și boala Canavan. Boala Krabbe și leucodistrofia metacromatică pot prezenta, de asemenea, un ADC scăzut în unele leziuni ale substanței albe, deoarece în timpul fazei acute de demielinizare poate apărea edemul intramielinic.
- Van der Knaap MS, Valk J. Classification of myelin disorders. În Van der Knaap MS, Valk J, eds. Rezonanța magnetică a mielinizării și a tulburărilor de mielină. Ed. a 3-a. Berlin: Springer, 2005, 20-24.
- Schiffmann R, van der Knaap MS. O abordare bazată pe RMN pentru diagnosticarea tulburărilor de substanță albă. Neurology 2009; 72: 750-759
- Takanashi J. Diagnostic imagistic al tulburărilor de substanță albă. Journal of the Japan Pediatric Society 2007; 111: 1243-1254.
- Van der Knaap MS, Breiter SN, Naidu S, et al. Definirea și clasificarea leucoencefalopatiilor de origine necunoscută: MR imaging approach. Radiology 1999; 213: 121-133.
.