Diagnóstico por imagem das anomalias da matéria branca | Hipomielinização cerebral congénita; rede para a doença de Pelizaeus-Merzbacher e doenças relacionadas
Diagnóstico por Imagem das Anormalidades da Matéria Branca
Junichi TAKANASHI, Departamento de Pediatria, Tokyo Women’s Medical University, Yachiyo Medical Center
Introdução
Neste trabalho, delineio a abordagem empregada para o diagnóstico de distúrbios que aparecem como sinais anormais na matéria branca cerebral em ressonância magnética (RM), desde a imagem até o diagnóstico. Os distúrbios que afetam principalmente a matéria branca são geralmente referidos como “leucoencefalopatia” ou “desordens da matéria branca” em inglês.1, 2) 3) Outro termo, leucodistrofia, às vezes é confundido com degeneração da matéria branca, mas isso na verdade se refere a um espectro mais restrito de distúrbios com um componente genético (distúrbios desmielinizantes herdados).
Classificação baseada em imagens dos distúrbios da matéria branca
O advento da RM melhorou drasticamente nossa capacidade de detectar lesões na matéria branca do sistema nervoso central. Muitas formas conhecidas de distúrbios da matéria branca exibem sinais específicos na RM, o que é útil para o seu diagnóstico. A identificação de padrões de anormalidade de matéria branca observada na RM (T1, T2 ou FLAIR) facilita o estreitamento das possibilidades em múltiplos diagnósticos diferenciais. A classificação de Schiffmann e van der Kamp das anormalidades da matéria branca de acordo com os achados da RM é de valor prático2, 4) (Figura 1, Tabela 1). Mesmo que isso não leve a um diagnóstico final, a classificação dos achados de imagem pode levar à descoberta posterior de um novo distúrbio. Aqui, descrevo os distúrbios de matéria branca em termos das classificações de RM acima, e explico os principais tipos de distúrbios.
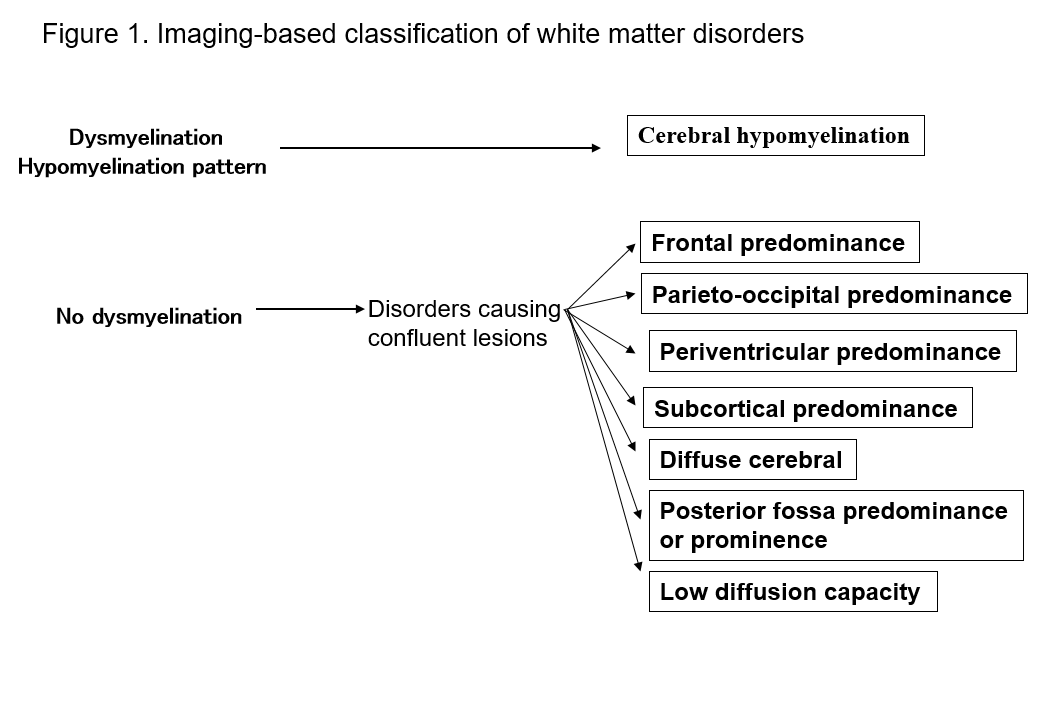
Tabela 1. Lista de desordens por padrões de RM
- Predomínio frontal
Doença de Alexander, variante frontal da adrenoleucodistrofia ligada ao X (ALD), leucodistrofia metacromática (MLD), leucodistrofia neuroaxonal com esferoides. - Predominância parieto-occipital
Adrenoleucodistrofia ligada ao X (ALD), doença de Krabbe, distúrbios peroxisomais de início precoce, hipoglicémia neonatal. - Predomínio periventricular
Leucodistrofia metacrômica (MLD), doença de Krabbe, síndrome de Sjögren-Larsson, doença corporal poliglucosana do adulto, leucoencefalopatia com envolvimento do tronco cerebral e medula espinhal e elevação do lactato (LBSL), leucomalácia periventricular (PVL), encefalopatia HIV, lipofuscinoses ceroidais neuronais de início tardio. - Predomínio subcortical
L-2-hydroxyglutaric aciduria, galactosemia, síndrome de Kearns-Sayer, academia propiônica, distúrbios do ciclo da uréia, doença de Canavan. - Cérebro difuso
Leucoencefalopatia megalencefálica com cistos subcorticais (MLC), leucoencefalopatia com desaparição de matéria branca (VWM), distrofia muscular congênita deficiente em merosina, doença mitocondrial, deficiência de cofactor de molibdénio, deficiência de sulfito oxidase, casos avançados de desordens da matéria branca. - Predomínio ou proeminência da fossa posterior
Leões do cerebelo e pedúnculos cerebelares: xantomatose cerebrotendinosa (CTX), distúrbios peroxisomais, doença de Alexander, leucoencefalopatia com envolvimento do tronco cerebral e medula espinhal e elevação do lactato (LBSL), doença do xarope de bordo, histiocitose, leucodistrofia autossômica dominante em adultos relacionada a uma duplicação da lamina B1, toxicidade da heroína e da cocaína.
Lesões do tronco encefálico: Doença de Alexander, LSBL, distúrbios peroxisomais, doença de Wilson, doença de Polyglucosan no adulto, síndrome de Leigh, atrofia dentatorubropallidoluysian (DRPLA), doença corporal de Polyglucosan no adulto, leucodistrofia autossômica dominante no adulto relacionada a uma duplicação da lamina B1. - Lesões multifocais
Síndrome de TORCH (infecção congênita por citomegalovírus), brucelose, encefalomielite aguda disseminada (ADEM), esclerose múltipla (EM), neuromielite óptica (NMO), arteriopatia autossômica dominante cerebral com infartos subcorticais e leucoencefalopatia (CADASIL), aterosclerose, angiopatia amilóide, doença cerebral associada à COL4A1, doença de Fabry, síndrome de Susac, doença mitocondrial, acidúria L-2-hidroxiglutarica, mucopolissacaridose (MPS), anormalidades cromossômicas (como a síndrome 6p-syndrome). - Lesões com baixa capacidade de difusão
Doença de urina em xarope de mármore, deficiência de metionina adenosiltransferase I/III, fenilcetonúria, hiperglicemia não-cetótica, doença de Canavan, lesões ativas na doença de Krabbe e leucodistrofia metacromática.
1. Hipomielinização da matéria branca cerebral
Refere-se a um grupo de distúrbios em que a formação da bainha da mielina é prejudicada ou retardada, e suas imagens se assemelham às de recém-nascidos com mielinização imatura. Nas imagens ponderadas em T2, a matéria branca aparece caracteristicamente como uma hiperintensidade generalizada que é ténue em comparação com o córtex. Para mais detalhes, veja o site da hipomielinização cerebral congênita.
Se as lesões de matéria branca não são consistentes com a hipomielinização cerebral de matéria branca, deve-se determinar se são confluentes ou múltiplas.2) As lesões de matéria branca confluentes são geralmente devidas à degeneração da matéria branca hereditária (leucodistrofia) e na maioria dos casos são bilateralmente simétricas. Lesões múltiplas de matéria branca são geralmente assimétricas e adquiridas. As lesões de matéria branca confluentes são ainda subdivididas nas categorias 2-7 abaixo.
2. Predomínio frontal
Neste grupo de desordens, lesões extensas de matéria branca estão presentes predominantemente no lobo frontal. Elas incluem doença de Alexander, a variante frontal da adrenoleucodistrofia ligada ao X (ALD), leucodistrofia metacromática (MLD), e leucodistrofia neuroaxonal com esferóides.
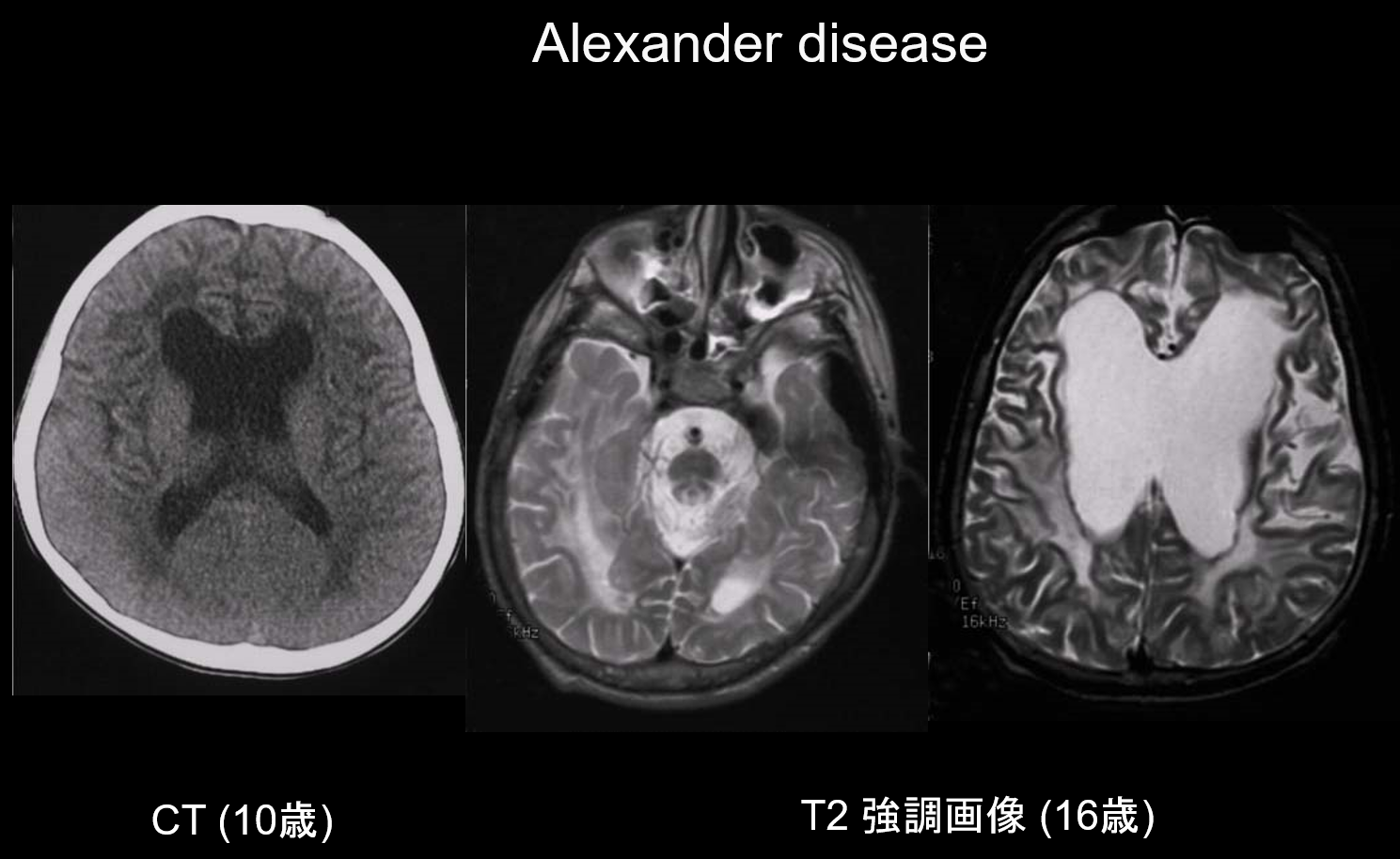
(a) doença de Alexander.
Doença de Alexander é uma desordem hereditária autossômica dominante causada por uma mutação no gene GFAP no cromossomo 17q21. Ela resulta no acúmulo de fibras de Rosenthal nas células gliais estreladas. Essas fibras são compostas de GFAP e proteínas de estresse (αB-crystallin e HSP27. A doença de Alexander ocorre principalmente na infância, entre 3 meses e 2 anos de idade, com aparecimento de megalencefalia, retardo de desenvolvimento, paralisia espástica e epilepsia. Na RM pode apresentar (i) lesões disseminadas de matéria branca, predominantemente no lobo frontal; (ii) marginação T1 hiperintensa e hipointensa T2 ao redor dos ventrículos laterais; (iii) lesões nos gânglios basais e tálamo; (iv) lesões no tronco encefálico; e (v) realce por contraste das lesões ativas (Figura 2). Nos estágios iniciais, juntamente com as lesões de matéria branca e putamen, observa-se inchaço que pode gradualmente causar atrofia ou formação de cisto.
3. Predomínio parieto-occipital
A principal característica deste grupo de distúrbios é a lesão da matéria branca parieto-occipital. Elas incluem adrenoleucodistrofia ligada ao X (ALD), doença de Krabbe, distúrbios peroxisômicos de início precoce e hipoglicemia neonatal.
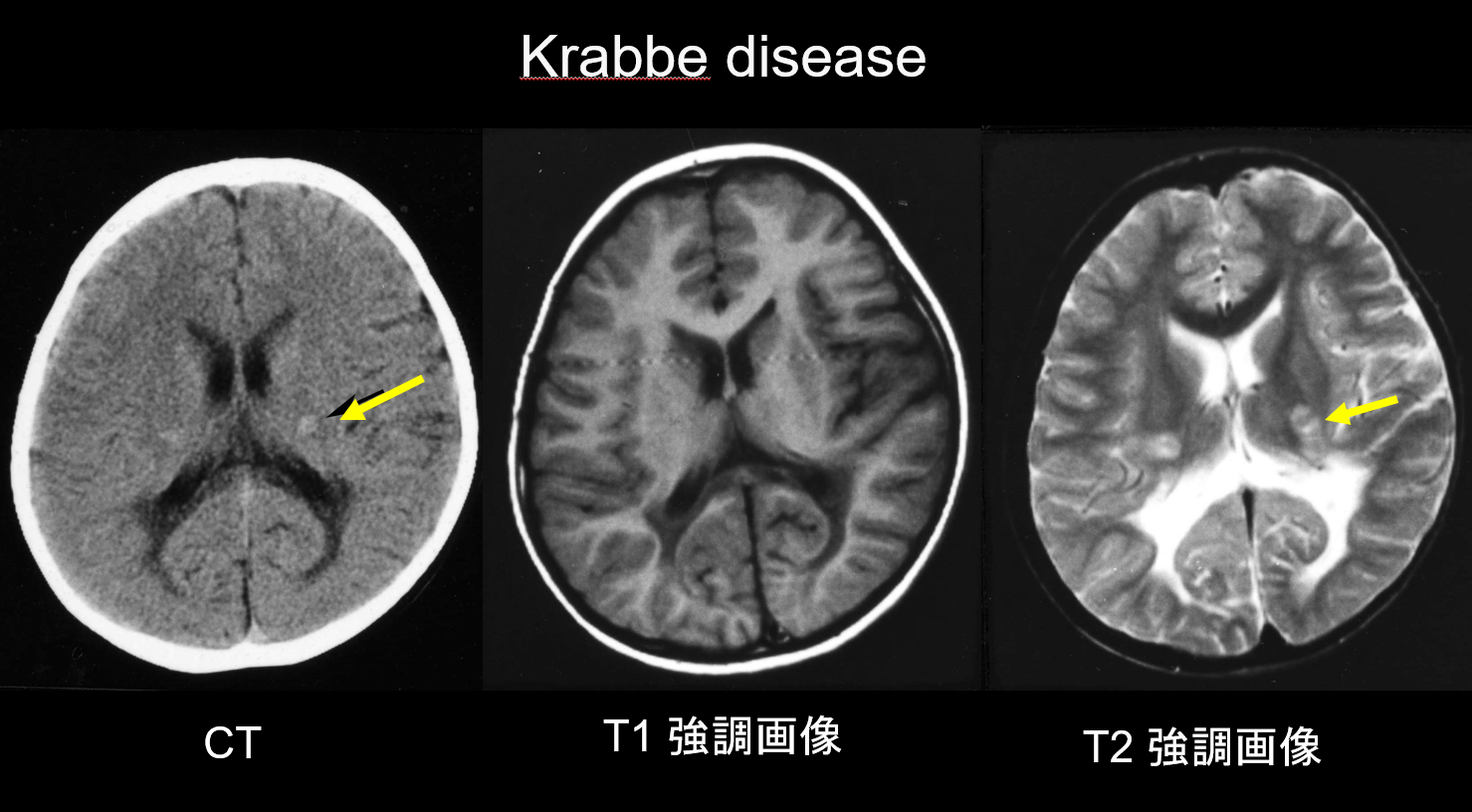
(a) Doença de Krabbe.
Krabbe disease is an autosomal recessive inherited disorder (lysosomal storage disease) caused by galactosylceramidase deficiency (chromosome 14q31), in which the accumulation of highly cytotoxic psychosine is believed to cause widespread demyelination. Também aparecem células grandes e multinucleadas chamadas “células globóides”. Dependendo da idade em que aparece, é classificada como doença infantil, de início de vida tardio, juvenil ou de início de vida adulta. A maioria dos casos são infantis e começam com o aparecimento de febre, irritabilidade, dificuldade de alimentação, atraso no desenvolvimento, neuropatia periférica, espasticidade e atrofia do nervo óptico aos 3-6 meses de idade. Durante os estágios iniciais, a tomografia computadorizada (TC) revela hiperdensidade característica no tálamo e na corona radiata. Acredita-se que isso reflete células globóides de alta densidade e proliferação glial. A RM também pode mostrar hiperintensidade T1 e hipointensidade T2 ao redor dos ventrículos, assim como estruturas lineares semelhantes às observadas na MLD (Figura 3). O núcleo cerebelar dentado, a matéria branca cerebelar e o trato piramidal do tronco encefálico apresentam hiperintensidade T2 desde um estágio inicial.
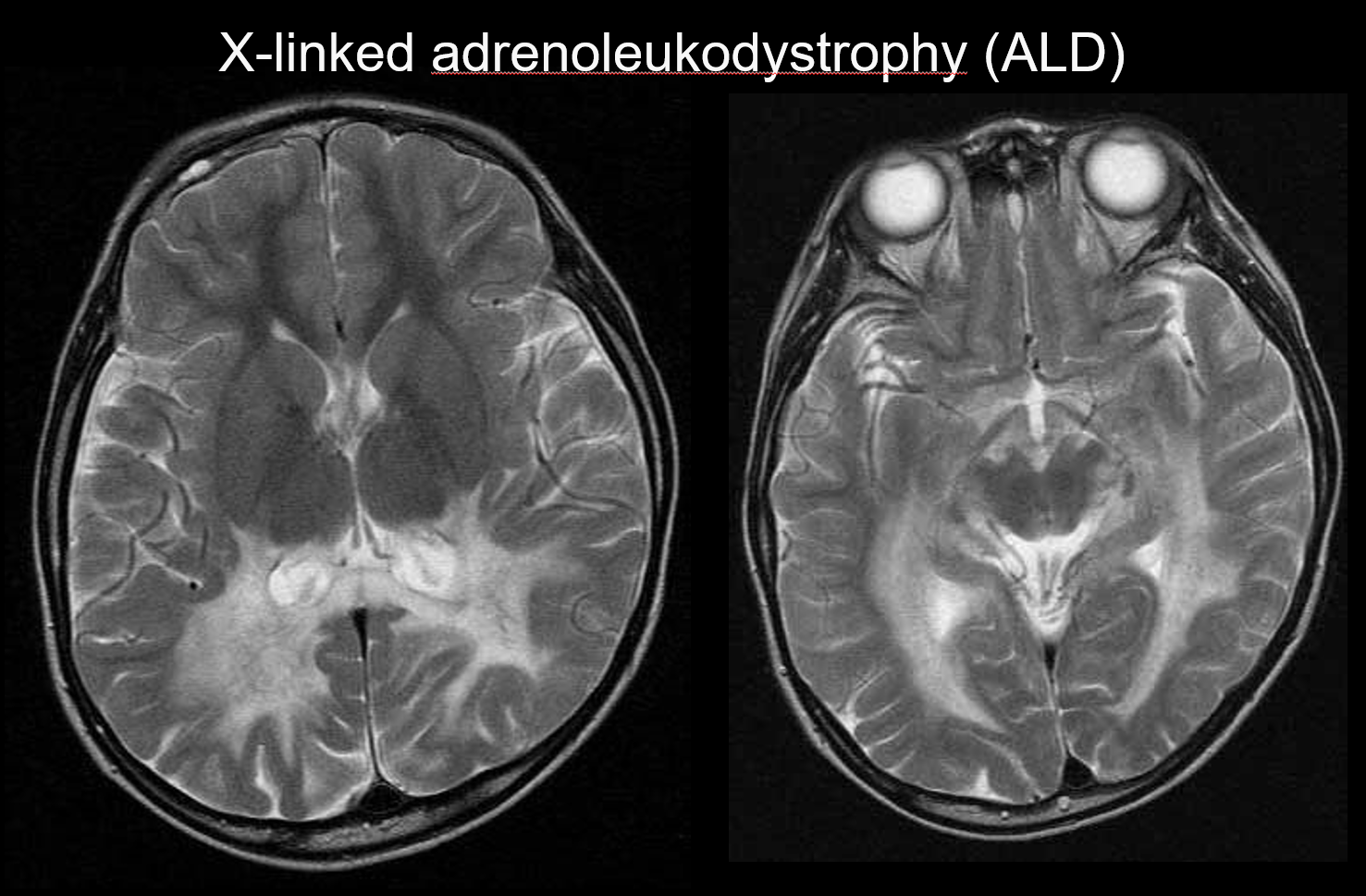
(b) Adrenoleucodistrofia ligada ao X
Adrenoleucodistrofia ligada ao X (ALD) é uma desordem hereditária recessiva ligada ao X (desordem peroxisomal) causada por uma anormalidade do gene ABCD1 (cromossomo Xq28). A oxidação deficiente do β resulta no acúmulo de ácidos graxos de cadeia muito longa na matéria branca cerebral e nas glândulas supra-renais, causando desmielinização e insuficiência adrenal. A ALD é categorizada em formas cerebrais infantis, adolescentes e adultos, adrenomieloneuropatia (AMN), e doença de Addison apenas. A forma cerebral infantil desenvolve-se aos 5-8 anos de idade, com o aparecimento de sintomas que incluem deterioração intelectual, marcha espástica e deficiência visual e auditiva. Patologicamente, a desmielinização progride da matéria branca que envolve o trígono do ventrículo lateral para o esplenium do corpus callosum, estendendo-se gradualmente em sentido anterolateral. Refletindo a patologia da doença, as hipersensibilidades simétricas em T2 e T1 que se estendem anterolateralmente da matéria branca que envolve o trígono do ventrículo lateral são aparentes na RM, com realce de contraste evidente nas margens (Figura 4). Lesões do trato corticospinal também são evidentes.
4. Predomínio periventricular
Estas alterações são caracterizadas principalmente por lesões na matéria branca que circunda os ventrículos laterais, com a matéria branca subcortical (fibras em U) sendo preservada. Este padrão é observado em numerosas desordens, incluindo a MLD, e é, portanto, comparativamente não específica. Sinais levemente anormais ao redor dos ventrículos laterais também são observados na degeneração cortical, particularmente nas lipofuscinoses ceróides neuronais que desenvolvem pós-infância.
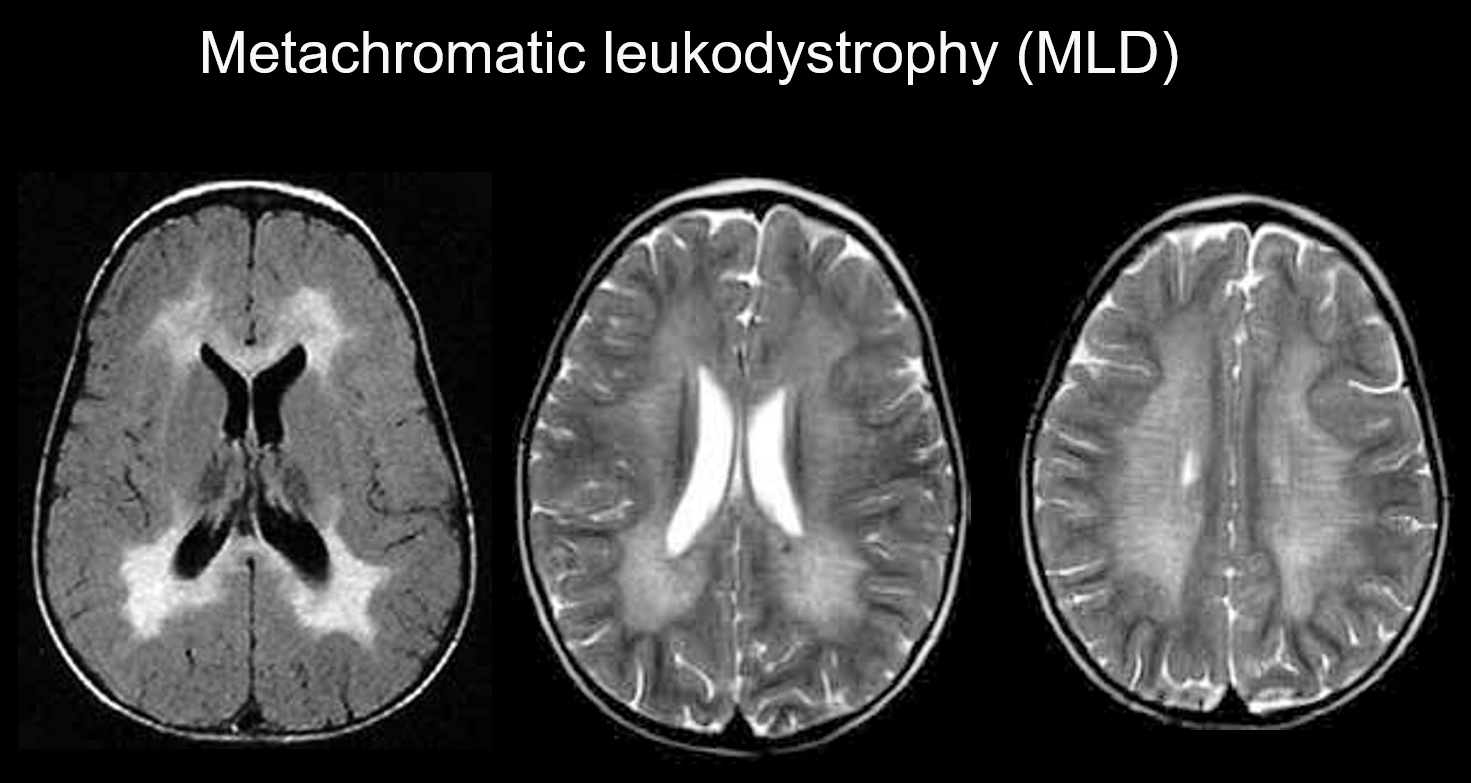
(a) Leucodistrofia metacromática.
Leucodistrofia metacromática é um distúrbio hereditário autossômico recessivo (distúrbio de armazenamento lisossômico) causado por deficiência de arilsulfatase A (cromossomo 22q13.31), no qual o acúmulo de sulfato altamente tóxico resulta em desmielinização. Dependendo da idade em que aparece, é classificada como congénita, infantil, juvenil ou adulta. Os seus sintomas incluem regressão cognitiva, paralisia espástica, movimentos involuntários, neuropatia periférica e atrofia do nervo óptico. Aparece nas imagens ponderadas em T2 como hiperintensidades da matéria branca principalmente ao redor dos ventrículos laterais, e nas imagens ponderadas em T1 como hipointensidades leves. As lesões tendem a ser predominantemente no lobo frontal. Bandas de intensidade normal (listras de tigre) podem ser evidentes dentro dos sinais anormais disseminados na matéria branca (Figura 5). Acredita-se que isto seja devido à preservação parcial da bainha de mielina no espaço perivascular e ao acúmulo de produtos de ruptura da bainha de mielina em macrófagos.
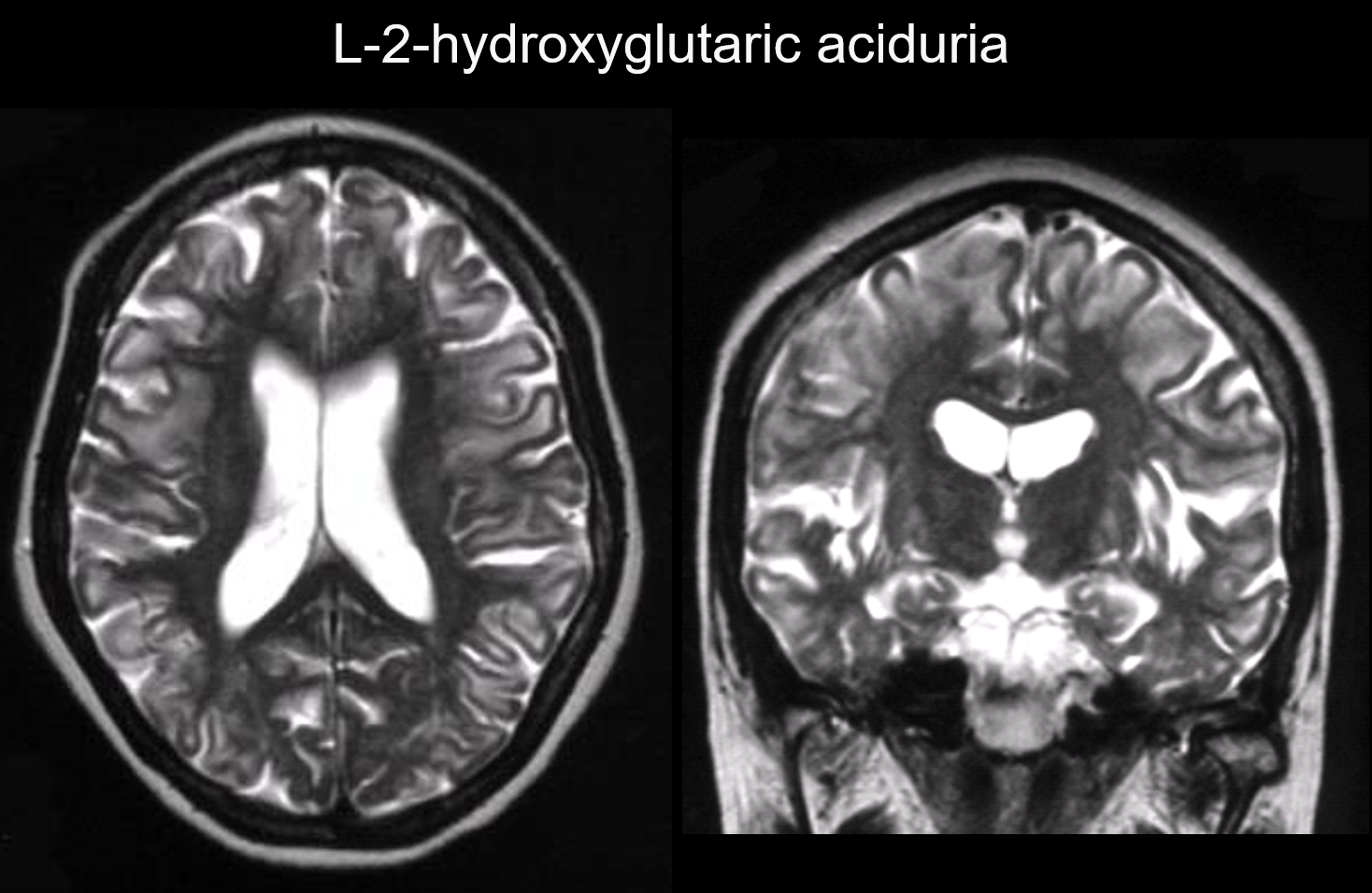
5. Predomínio subcortical
Nestas perturbações, as lesões ocorrem principalmente na matéria branca subcortical, incluindo as fibras U. Distúrbios com este padrão incluem a acidúria L-2-hidroxi-glutarica (Figura 6), galactosemia, síndrome de Kearns-Sayer, academia propiônica, distúrbios do ciclo da uréia e doença de Canavan em estágio inicial.
6. Cérebro difuso
Nesses distúrbios, sinais anormais aparecem em toda a matéria branca cerebral. Elas exibem fortes hiperintensidades T2 em relação aos sinais T2 produzidos pela matéria branca não mielinizada (hipomielinização). Além dos casos de leucoencefalopatia megalencefálica com cistos subcorticais e leucoencefalopatia com desaparição da matéria branca, pacientes com qualquer tipo de distúrbio de matéria branca eventualmente exibem esse padrão à medida que a doença avança.
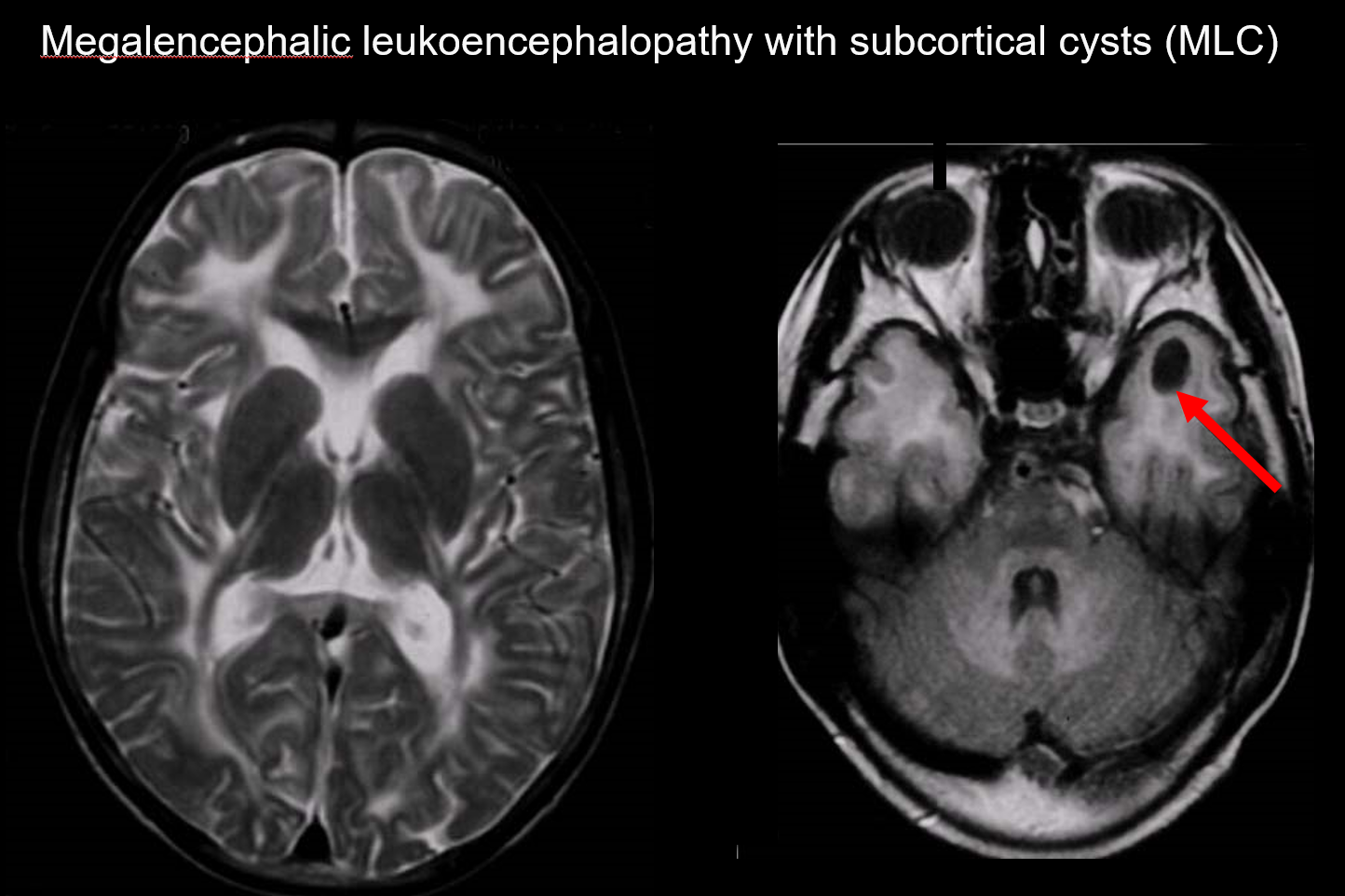
(a) Leucoencefalopatia megalencefálica com cistos subcorticais (MLC)
MLC é um distúrbio hereditário autossômico recessivo causado por uma anormalidade do gene MLC1, e seu início na infância é marcado por megalocefalia, progressão lenta da deterioração motora, ataxia e espasticidade. A RM revela sinais anormais característicos disseminados na matéria branca e leve edema da matéria branca, assim como a formação de cistos nos lobos parietais e temporais (Figura 7).7, 8) As imagens ponderadas em T1 e T2 revelam matéria branca anormal, enquanto os cistos apresentam todos hipointensidade T1 e hiperintensidade T2, tornando-os particularmente difíceis de detectar. A imagem FLAIR, que visualiza os quistos (água) como hipointensidades, é valiosa para o seu diagnóstico. É mais comum entre os japoneses do que a leucoencefalopatia megalencefálica vacuolante.
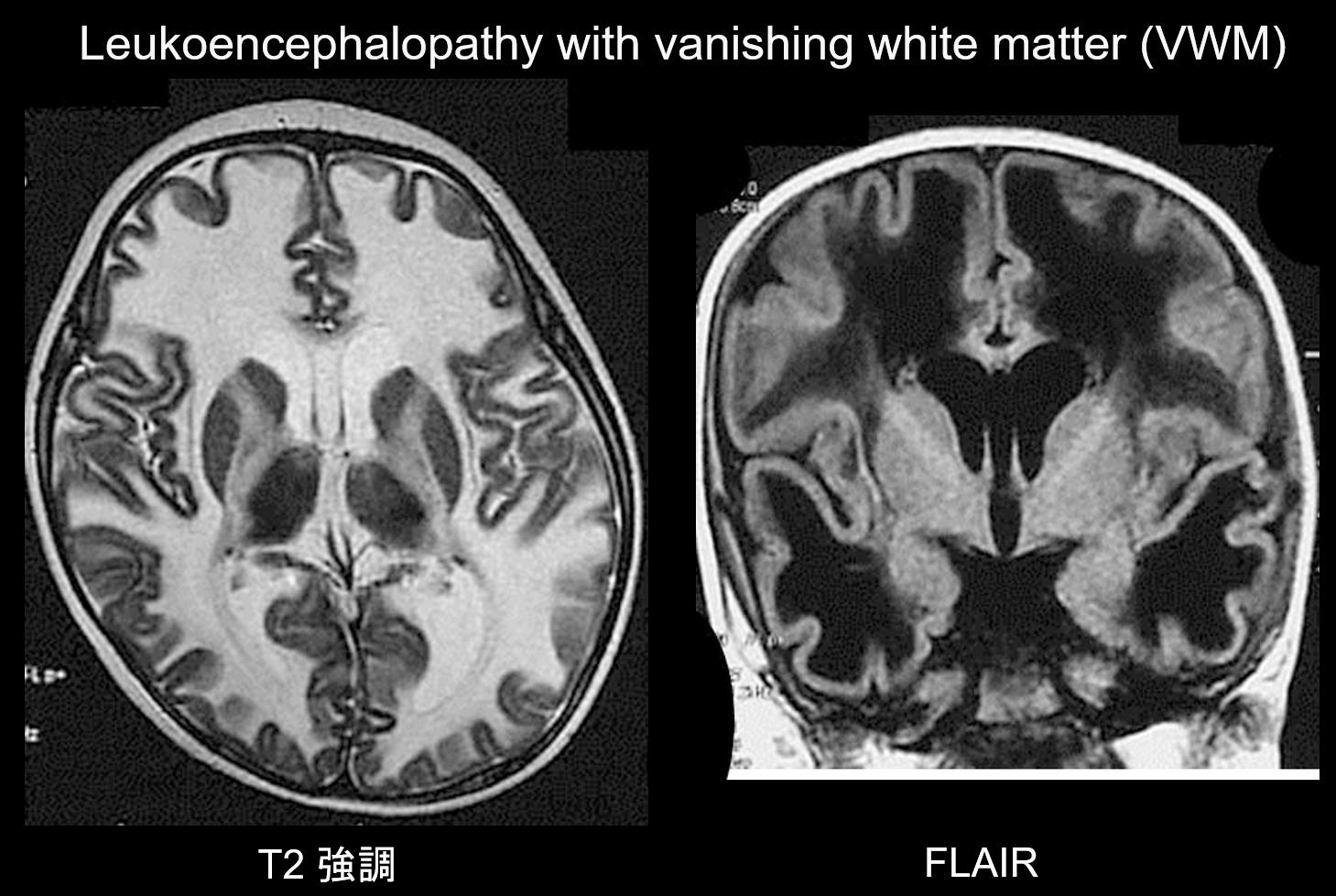
(b) Leucoencefalopatia com desaparição da matéria branca.
Leucoencefalopatia com matéria branca desaparecida (MVA) é um distúrbio autossômico recessivo herdado causado por uma deficiência de eIF2B, uma proteína associada à eIF2, que transfere o RNAt iniciador para ribossomos. A eIF2B consiste de cinco proteínas diferentes, todas com loci genético diferente. O VWM demonstrou ser a mesma desordem que a ataxia cerebelar infantil e a hipomielinização central (CACH). Os pacientes são normais durante o período neonatal e na primeira infância, mas após a instalação (geralmente aos 2-6 anos de idade) eles desenvolvem lentamente regressão cognitiva progressiva, espasticidade e ataxia. Estes sintomas são conhecidos por serem exacerbados por infecções ou traumas menores. A matéria branca cerebral apresenta hiperintensidade T2 generalizada e hipointensidade T1 e é gradualmente substituída por fluido ao longo do tempo (como o nome indica, a matéria branca desaparece) (Figura 8). A matéria branca cística contém estruturas enfaixadas que se acredita representarem o tecido remanescente. Sinais anormais também são vistos no tronco cerebral, particularmente no trato tegmental central. A imagem FLAIR é valiosa para o diagnóstico desta doença.
7. Predomínio ou proeminência da fossa posterior
Estas desordens são caracterizadas por lesões predominantemente no tronco cerebral e no cerebelo. Lesões cerebelares de matéria branca podem ser causadas por desordens incluindo xantomatose cerebrotendinosa (CTX), desordens peroxisomais, doença de Alexander, leucoencefalopatia com envolvimento do tronco cerebral e medula espinhal e elevação do lactato (LBSL), doença da urina de xarope de bordo, histiocitose, e toxicidade da heroína e cocaína. As lesões do tronco cerebral podem ser causadas por distúrbios como a doença de Alexander, LSBL e a doença de poliglucosan no adulto. Lesões do pedúnculo cerebelar médio são observadas na síndrome do X frágil e na leucodistrofia autossômica dominante do adulto relacionada a uma duplicação da lâmina B1.
8. Lesões multifocais
Desordens não semelhantes que produzem as lesões confluentes descritas em 2-7 acima, as desordens nesta seção resultam em lesões multifocais (dispersas). Elas incluem infecções como a síndrome de TORCH (devido a infecção congênita por citomegalovírus ou outra causa) e brucelose; distúrbios inflamatórios como a encefalomielite aguda disseminada (ADEM), esclerose múltipla (EM) e a neuromielite óptica (NMO); vasculopatias como a arteriopatia cerebral autossômica dominante com infartos subcorticais e leucoencefalopatia (CADASIL), aterosclerose, angiopatia amilóide, doença cerebral associada à COL4A1, doença de Fabry e síndrome de Susac; e condições hereditárias como doença mitocondrial, acidúria L-2-hydroxyglutaric aciduria, mucopolissacaridose (MPS), e anormalidades cromossômicas (como a síndrome 6p-syndrome).
9. Lesões com baixa capacidade de difusão
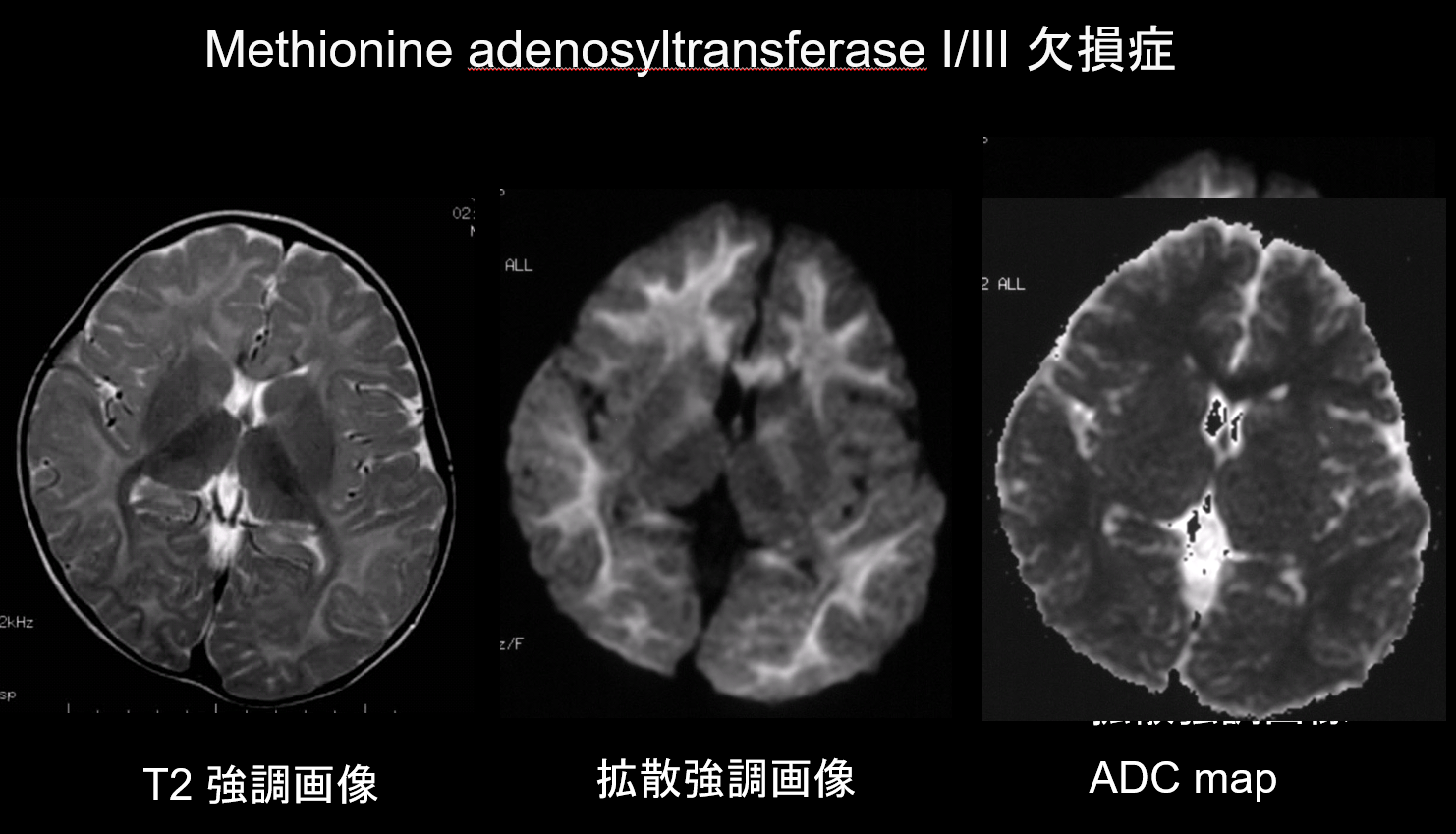
Tanto na desmielinização quanto na hipomielinização, as principais patologias das doenças da matéria branca, há uma diminuição da quantidade de mielina, o que limita a difusão, e o correspondente aumento do líquido extracelular resulta em hiperintensidades em T2 com um alto coeficiente de difusão aparente (DCV). É raro que as doenças de matéria branca exibam tanto hiperintensidades T2 quanto baixo ADC, e essa combinação é, portanto, de alto valor diagnóstico. Os distúrbios caracterizados pela presença de edema intramielínico na bainha da mielina e nos espaços entre as bainhas exibem um baixo ADC. Eles incluem doença de urina de xarope de bordo, deficiência de metionina adenosiltransferase I/III (Figura 9), fenilcetonúria, hiperglicemia não-cetótica, e doença de Canavan. A doença de Krabbe e a leucodistrofia metacromática também podem apresentar baixo ADC em algumas lesões de matéria branca, já que o edema intramielínico pode ocorrer durante a fase aguda de desmielinização.
- Van der Knaap MS, Valk J. Classification of myelin disorders. Em Van der Knaap MS, Valk J, eds. Ressonância magnética de mielinização e distúrbios mielínicos. 3ª ed. Berlim: Springer, 2005, 20-24.
- Schiffmann R, van der Knaap MS. Uma abordagem baseada em ressonância magnética para o diagnóstico de distúrbios da matéria branca. Neurologia 2009; 72: 750-759
- Takanashi J. Diagnostic imaging of white matter disorders. Journal of the Japan Pediatric Society 2007; 111: 1243-1254.
- Van der Knaap MS, Breiter SN, Naidu S, et al. Definindo e categorizando leucoencefalopatias de origem desconhecida: Abordagem de imagens MR. Radiologia 1999; 213: 121-133.