Diagnostic Imaging of White Matter Abnormalities | Congenitale cerebrale hypomyelinisatie; netwerk voor de ziekte van Pelizaeus-Merzbacher en aanverwante aandoeningen
Diagnostic Imaging of White Matter Abnormalities
Junichi TAKANASHI, Afdeling Kindergeneeskunde, Tokyo Women’s Medical University, Yachiyo Medical Center
Inleiding
In dit artikel schets ik de aanpak die wordt gebruikt voor de diagnose van aandoeningen die verschijnen als abnormale signalen in de witte hersenstof op magnetische resonantie beeldvorming (MRI), van beeldvorming tot diagnose. Aandoeningen die voornamelijk de witte stof aantasten worden in het Engels meestal “leukoencephalopathy” of “white matter disorders” genoemd.1, 2) 3) Een andere term, leukodystrofie, wordt soms verward met witte stof degeneratie, maar dit verwijst eigenlijk naar een beperkter spectrum van aandoeningen met een genetische component (erfelijke demyeliniserende aandoeningen).
Imaging-based classificatie van witte stof aandoeningen
De komst van MRI heeft ons vermogen om laesies in de witte stof van het centrale zenuwstelsel op te sporen drastisch verbeterd. Vele bekende vormen van witte stof afwijkingen vertonen specifieke tekens op MRI, wat nuttig is voor hun diagnose. Het identificeren van patronen van witte stof abnormaliteit gezien op MRI (T1-gewogen, T2-gewogen, of FLAIR beeldvorming) maakt het gemakkelijk om de mogelijkheden in meerdere differentiële diagnoses te beperken. Schiffmann en van der Kamp’s classificatie van witte stof afwijkingen op basis van MRI bevindingen is van praktische waarde2, 4) (figuur 1, tabel 1). Zelfs als dit niet leidt tot een definitieve diagnose, kan de classificatie van beeldvormende bevindingen leiden tot de latere ontdekking van een nieuwe aandoening. Hier beschrijf ik witte stof stoornissen in termen van de bovenstaande MRI-classificaties, en verklaar de belangrijkste soorten stoornissen.
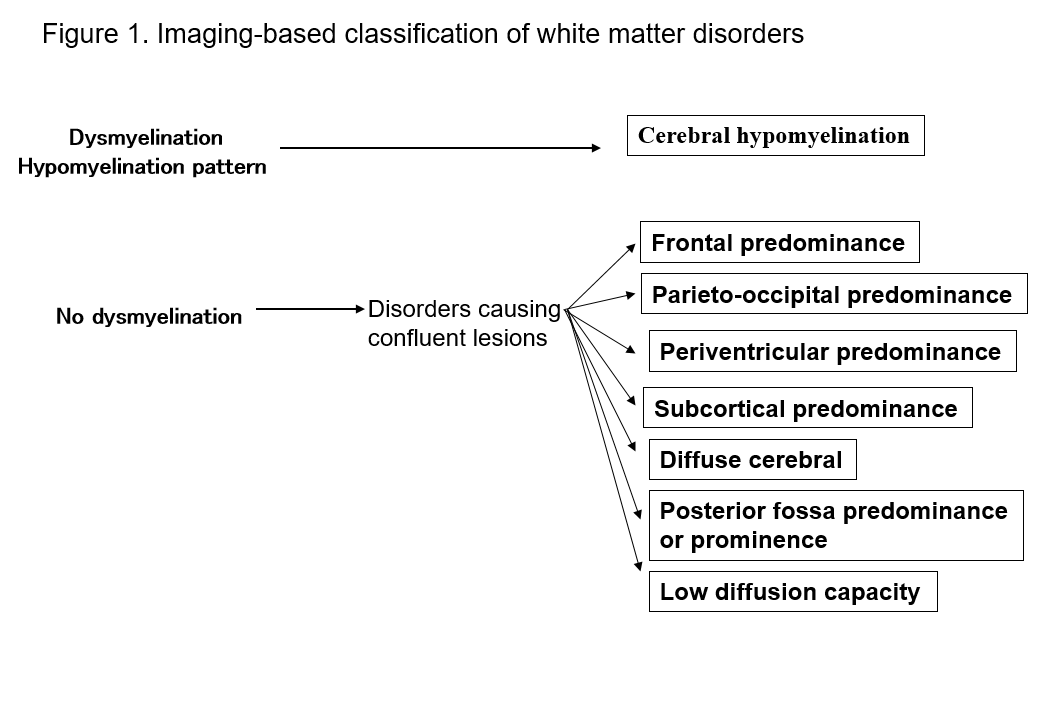
Tabel 1. Lijst van aandoeningen naar MRI-patroon
- Frontale predominantie
De ziekte van Alexander, frontale variant van X-gebonden adrenoleukodystrofie (ALD), metachromatische leukodystrofie (MLD), neuroaxonale leukodystrofie met sferoïden. - Pariëto-occipitale predominantie
X-gebonden adrenoleukodystrofie (ALD), ziekte van Krabbe, vroeg optredende peroxisomale aandoeningen, neonatale hypoglykemie. - Periventriculaire predominantie
Metachromatische leukodystrofie (MLD), ziekte van Krabbe, syndroom van Sjögren-Larsson, polyglucosan-lichaampjesziekte bij volwassenen, leukoencefalopathie met betrokkenheid van hersenstam en ruggenmerg en lactaatstijging (LBSL), periventriculaire leukomalacie (PVL), HIV-encefalopathie, later ontstane neuronale ceroïde lipofuscinosen. - Subcorticale predominantie
L-2-hydroxyglutaarzuururie, galactosemie, Kearns-Sayer syndroom, propionzuur academie, ureum cyclus stoornissen, Canavan ziekte. - Diffuse cerebrale
Megalencephale leukoencephalopathie met subcorticale cysten (MLC), leukoencephalopathie met verdwijnende witte stof (VWM), merosine deficiënte congenitale spierdystrofie, mitochondriale ziekte, molybdeen cofactor deficiëntie, sulfiet oxidase deficiëntie, gevorderde gevallen van witte stof stoornissen. - Overheersing of prominentie van de posterieure fossa
Lesies van het cerebellum en de cerebellaire peduncles: cerebrotendineuze xanthomatose (CTX), peroxisomale aandoeningen, ziekte van Alexander, leukoencefalopathie met betrokkenheid van hersenstam en ruggenmerg en lactaatverhoging (LBSL), maple syrup urine disease, histiocytose, volwassen autosomaal dominante leukodystrofie gerelateerd aan een lamin B1 duplicatie, heroïne- en cocaïnegiftigheid.
Hersenstam laesies: Ziekte van Alexander, LSBL, peroxisomale aandoeningen, ziekte van Wilson, polyglucosaanziekte bij volwassenen, Leigh-syndroom, dentatorubropallidoluysische atrofie (DRPLA), polyglucosaanlichaamsziekte bij volwassenen, autosomaal dominante leukodystrofie bij volwassenen gerelateerd aan een lamin B1-duplicatie. - Multifocale laesies
TORCH-syndroom (congenitale cytomegalovirusinfectie), brucellose, acute gedissemineerde encefalomyelitis (ADEM), multiple sclerose (MS), neuromyelitis optica (NMO), cerebrale autosomaal-dominante arteriopathie met subcorticale infarcten en leukoencephalopathie (CADASIL), atherosclerose, amyloïde angiopathie, COL4A1-geassocieerde cerebrale small-vessel disease, ziekte van Fabry, Susac-syndroom, mitochondriale ziekte, L-2-hydroxyglutaarzuururie, mucopolysaccharidose (MPS), chromosoomafwijkingen (zoals 6p-syndroom). - Lesies met een lage diffusiecapaciteit
Maple syrup urine disease, methionine adenosyltransferase I/III-deficiëntie, fenylketonurie, non-ketotische hyperglycinemie, ziekte van Canavan, actieve lesies bij de ziekte van Krabbe, en metachromatische leukodystrofie.
1. Hypomyelinisatie van de witte hersenstof
Dit verwijst naar een groep van aandoeningen waarbij de vorming van de myelineschede verstoord of vertraagd is, en de beelden lijken op die van pasgeborenen met onvolgroeide myelinisatie. Op T2-gewogen beelden verschijnt de witte stof karakteristiek als een wijdverspreide hyperintensiteit die vaag is vergeleken met de cortex. Voor meer details, zie de website van congenitale cerebrale hypomyelinatio.
Als witte stof laesies niet consistent zijn met cerebrale witte stof hypomyelinisatie, moet worden bepaald of ze confluent of multipel zijn.2) Confluente witte stof laesies zijn meestal te wijten aan erfelijke witte stof degeneratie (leukodystrofie) en zijn in de meeste gevallen bilateraal symmetrisch. Meervoudige witte stof laesies zijn meestal asymmetrisch en verworven. Confluente witte stof laesies zijn verder onderverdeeld in de categorieën 2-7 hieronder.
2. Frontale predominantie
In deze groep van aandoeningen, uitgebreide witte stof laesies zijn aanwezig voornamelijk in de frontale kwab. Hiertoe behoren de ziekte van Alexander, de frontale variant van X-gebonden adrenoleukodystrofie (ALD), metachromatische leukodystrofie (MLD), en neuroaxonale leukodystrofie met sferoïden.
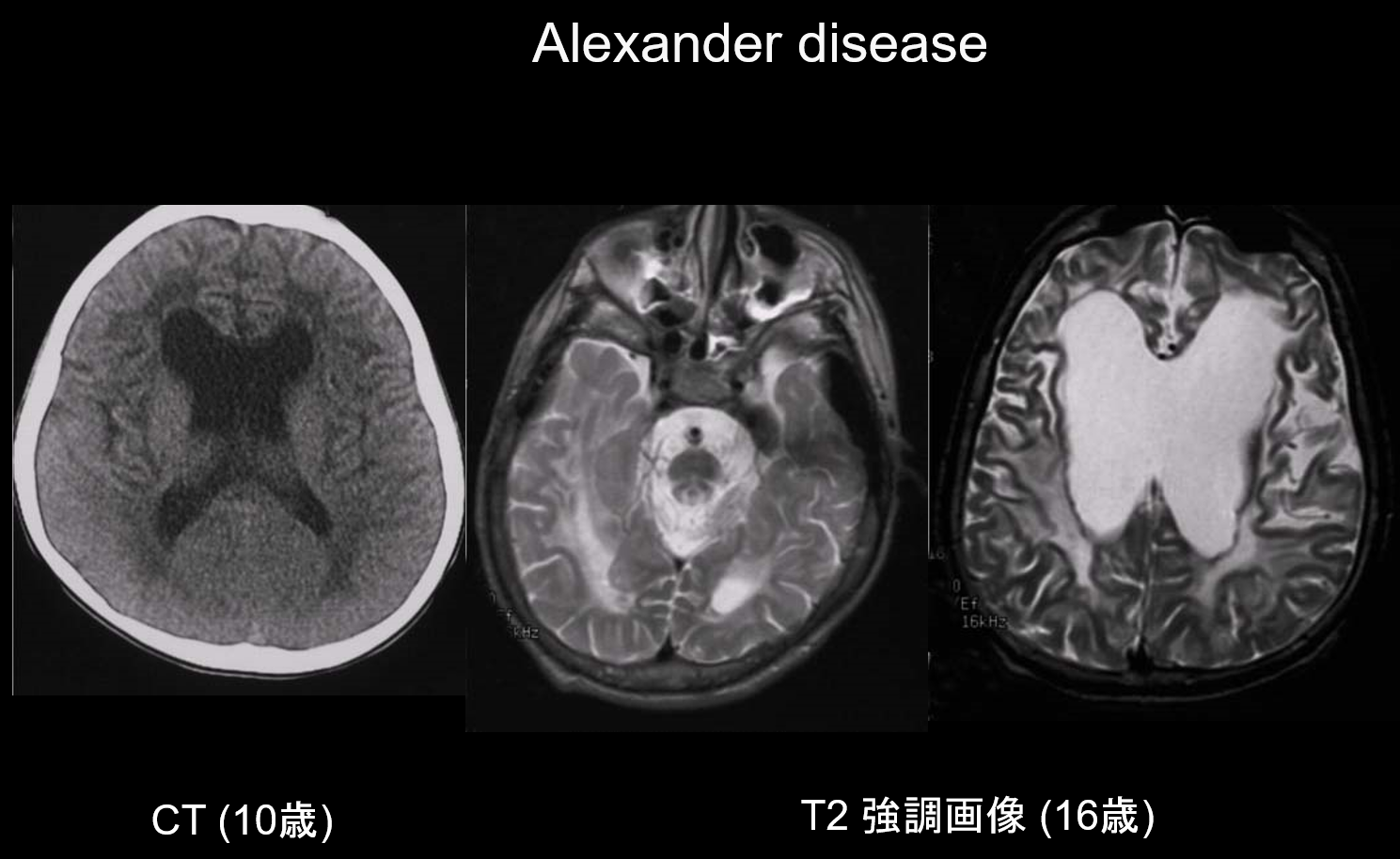
(a) Ziekte van Alexander.
De ziekte van Alexander is een autosomaal dominant overervende aandoening die wordt veroorzaakt door een mutatie in het GFAP-gen op chromosoom 17q21. Het resulteert in de accumulatie van Rosenthal vezels in de stellate gliacellen. Deze vezels zijn samengesteld uit GFAP en stress-eiwitten (αB-crystalline en HSP27. De ziekte van Alexander komt vooral voor in de zuigelingenleeftijd, tussen 3 maanden en 2 jaar, met megalencefalie, ontwikkelingsachterstand, spastische verlamming en epilepsie. Op MRI, kan vertonen (i) wijdverspreide witte stof laesies, voornamelijk in de frontale kwab, (ii) T1 hyperintense en T2 hypointense margination rond de laterale ventrikels, (iii) laesies in de basale ganglia en thalamus, (iv) hersenstam laesies, en (v) contrastversterking van actieve laesies (figuur 2). In de vroege stadia wordt samen met witte stof- en putamenlaesies zwelling gezien die geleidelijk atrofie of cystevorming kan veroorzaken.
3. Pariëto-occipitale predominantie
Het belangrijkste kenmerk van deze groep aandoeningen is pariëto-occipitale witte stof laesies. Zij omvatten X-gebonden adrenoleukodystrofie (ALD), de ziekte van Krabbe, peroxisomale aandoeningen van vroege datum, en neonatale hypoglykemie.
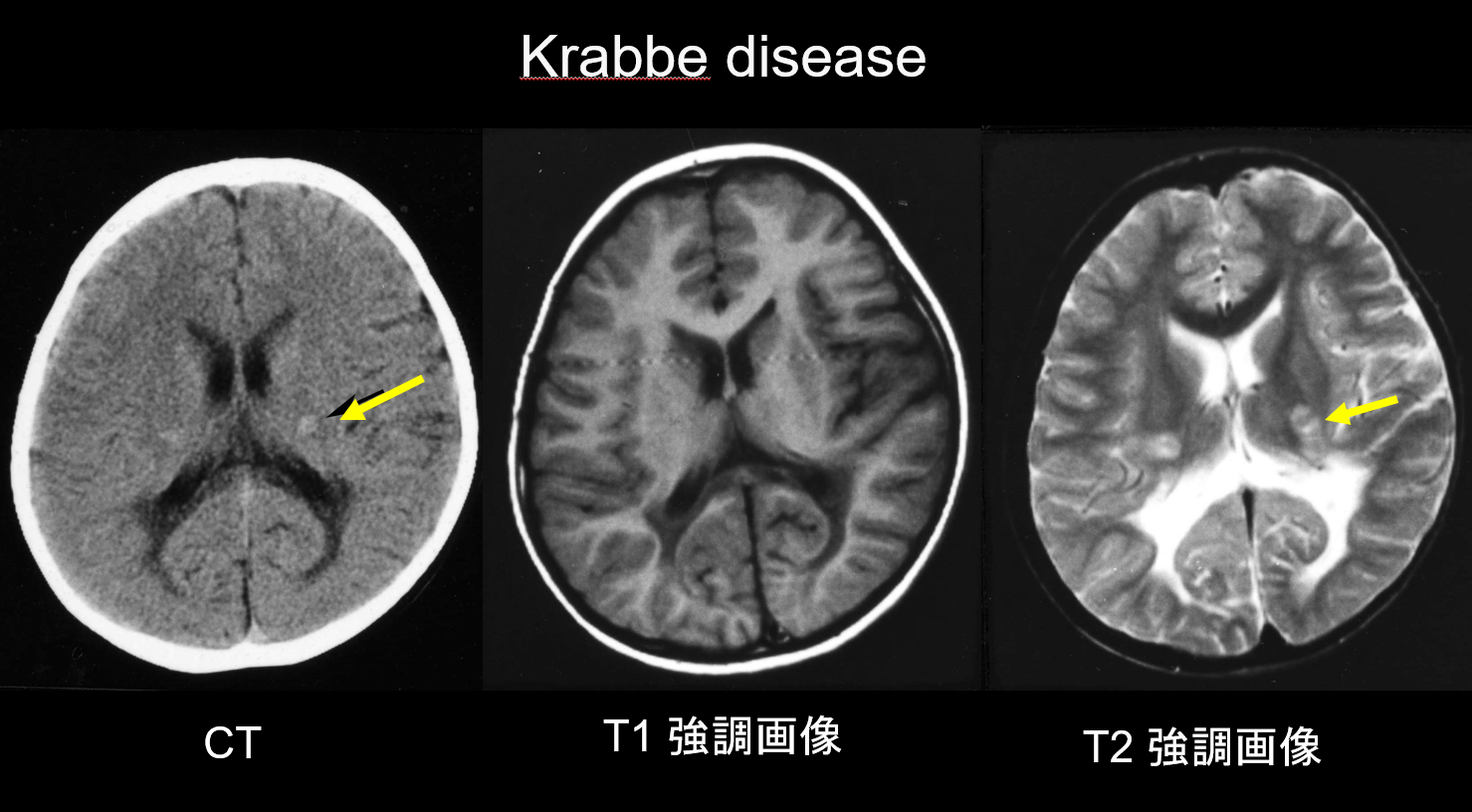
(a) Ziekte van Krabbe.
De ziekte van Krabbe is een autosomaal recessief overervende aandoening (lysosomale opslagziekte) veroorzaakt door galactosylceramidase-deficiëntie (chromosoom 14q31), waarbij de accumulatie van zeer cytotoxisch psychosine wordt verondersteld wijdverbreide demyelinisatie te veroorzaken. Ook verschijnen er grote, meercellige cellen die “globoïde cellen” worden genoemd. Afhankelijk van de leeftijd waarop de ziekte zich voordoet, wordt zij geclassificeerd als infantiel, laat-onset infantiel, juveniel-onset, of volwassen-onset ziekte. De meeste gevallen zijn infantiel en beginnen met het optreden van koorts, prikkelbaarheid, moeilijkheden met voeden, ontwikkelingsachterstand, perifere neuropathie, spasticiteit en atrofie van de oogzenuw op de leeftijd van 3-6 maanden. In het beginstadium toont computertomografie (CT) een karakteristieke hyperdensiteit in de thalamus en corona radiata. Dit wordt verondersteld een weerspiegeling te zijn van globoïde cellen met hoge dichtheid en gliale proliferatie. MRI kan ook T1 hyperintensiteit en T2 hypointensiteit rond de ventrikels vertonen, evenals lineaire structuren die lijken op die welke bij MLD worden gezien (figuur 3). De dentaatkern van de hersenen, de witte stof van de hersenen en de pyramidale tractus van de hersenstam vertonen al in een vroeg stadium T2-hyperintensiteit.
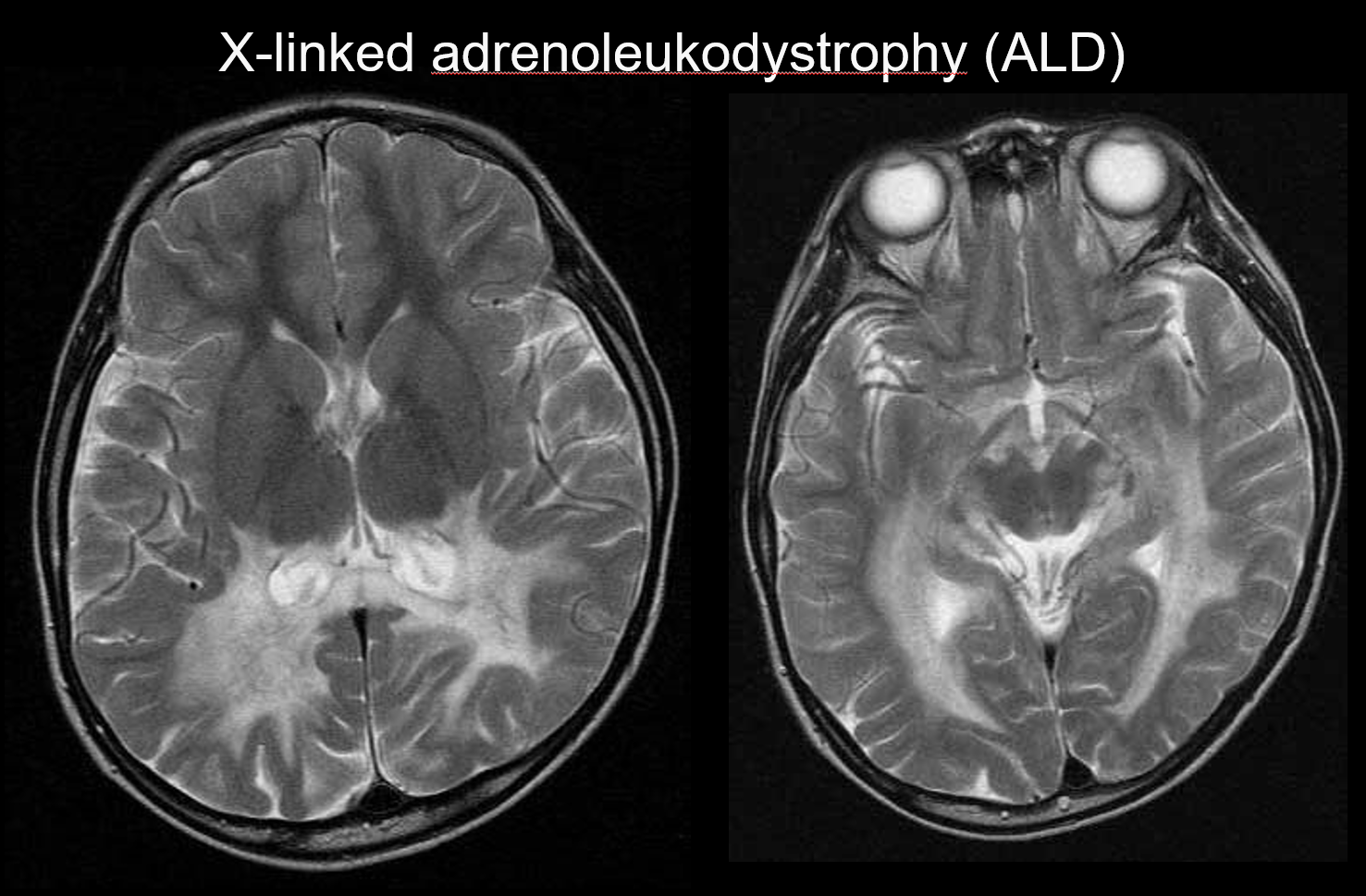
(b) X-gebonden adrenoleukodystrofie
X-gebonden adrenoleukodystrofie (ALD) is een X-gebonden recessief overervende aandoening (peroxisomale stoornis) veroorzaakt door een afwijking van het ABCD1-gen (chromosoom Xq28). Gestoorde β-oxidatie leidt tot accumulatie van zeer lange-keten vetzuren in de witte hersenstof en de bijnieren, wat demyelinisatie en bijnierinsufficiëntie tot gevolg heeft. ALD wordt ingedeeld in de kinder-, adolescentie- en volwassen cerebrale vorm, adrenomyeloneuropathie (AMN), en de ziekte van Addison alleen. De kinder cerebrale vorm ontwikkelt zich op de leeftijd van 5-8 jaar, met symptomen als intellectuele achteruitgang, spastische gang, en slechtziendheid en gehoorstoornissen. Pathologisch ontwikkelt de demyelinisatie zich van de witte stof rond de trigone van de laterale ventrikel tot het splenium van het corpus callosum, zich geleidelijk anterolateraal uitbreidend. De ziekte pathologie weerspiegelt symmetrische T2 hyperintensiteiten en T1 hypointensiteiten die zich anterolateraal uitstrekken van de witte stof rond de trigone van de laterale ventrikel zijn zichtbaar op MRI, met contrastversterking duidelijk aan de randen (figuur 4). Corticospinale tractus laesies zijn ook duidelijk.
4. Periventriculaire predominantie
Deze aandoeningen worden voornamelijk gekenmerkt door laesies in de witte stof rond de laterale ventrikels, waarbij de subcorticale witte stof (U-vezels) behouden blijft. Dit patroon wordt gezien bij talrijke aandoeningen, waaronder MLD, en is derhalve betrekkelijk niet-specifiek. Licht abnormale signalen rond de laterale ventrikels worden ook gezien bij corticale degeneratie, met name bij neuronale ceroïde lipofuscinosen die zich na de kindertijd ontwikkelen.
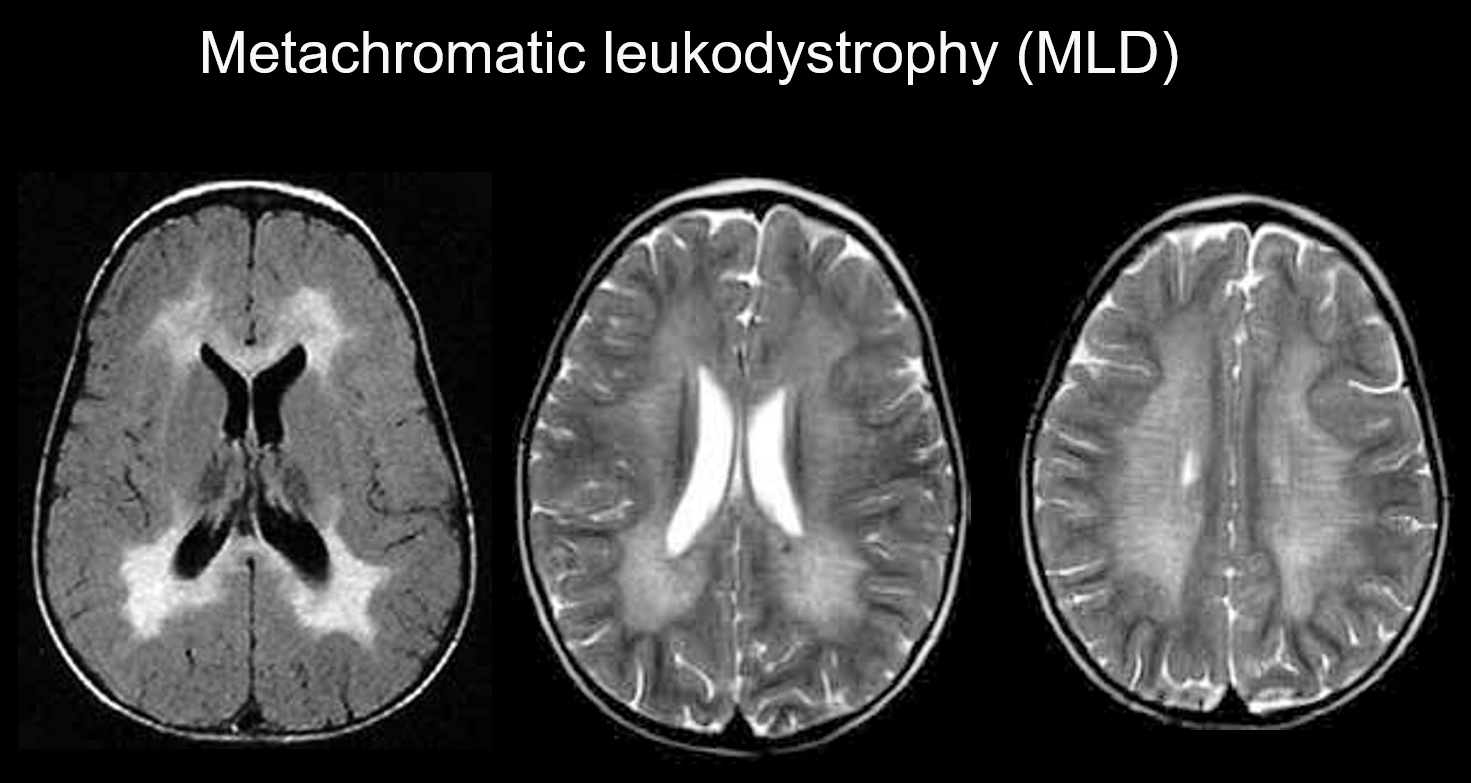
(a) Metachromatische leukodystrofie.
Metachromatische leukodystrofie is een autosomaal recessief overervende aandoening (lysosomale opslagstoornis) die wordt veroorzaakt door arylsulfatase-A-deficiëntie (chromosoom 22q13.31), waarbij de ophoping van zeer giftig sulfatide leidt tot demyelinisatie. Afhankelijk van de leeftijd waarop de ziekte zich manifesteert, wordt zij geclassificeerd als aangeboren, infantiel, juveniel of volwassen. De symptomen zijn onder meer cognitieve achteruitgang, spastische verlamming, onwillekeurige bewegingen, perifere neuropathie en atrofie van de oogzenuw. De aandoening verschijnt op T2-gewogen beeldvorming als hyperintensiteiten van de witte stof, voornamelijk rond de laterale ventrikels, en op T1-gewogen beeldvorming als milde hypointensiteiten. De letsels hebben de neiging zich hoofdzakelijk in de frontale kwab voor te doen. Binnen de wijdverspreide abnormale signalen in de witte stof kunnen banden van normale intensiteit (tijgerstrepen) zichtbaar zijn (figuur 5). Deze worden verondersteld te wijten te zijn aan het gedeeltelijke behoud van de myelineschede in de perivasculaire ruimte en aan de accumulatie van afbraakproducten van de myelineschede in macrofagen.
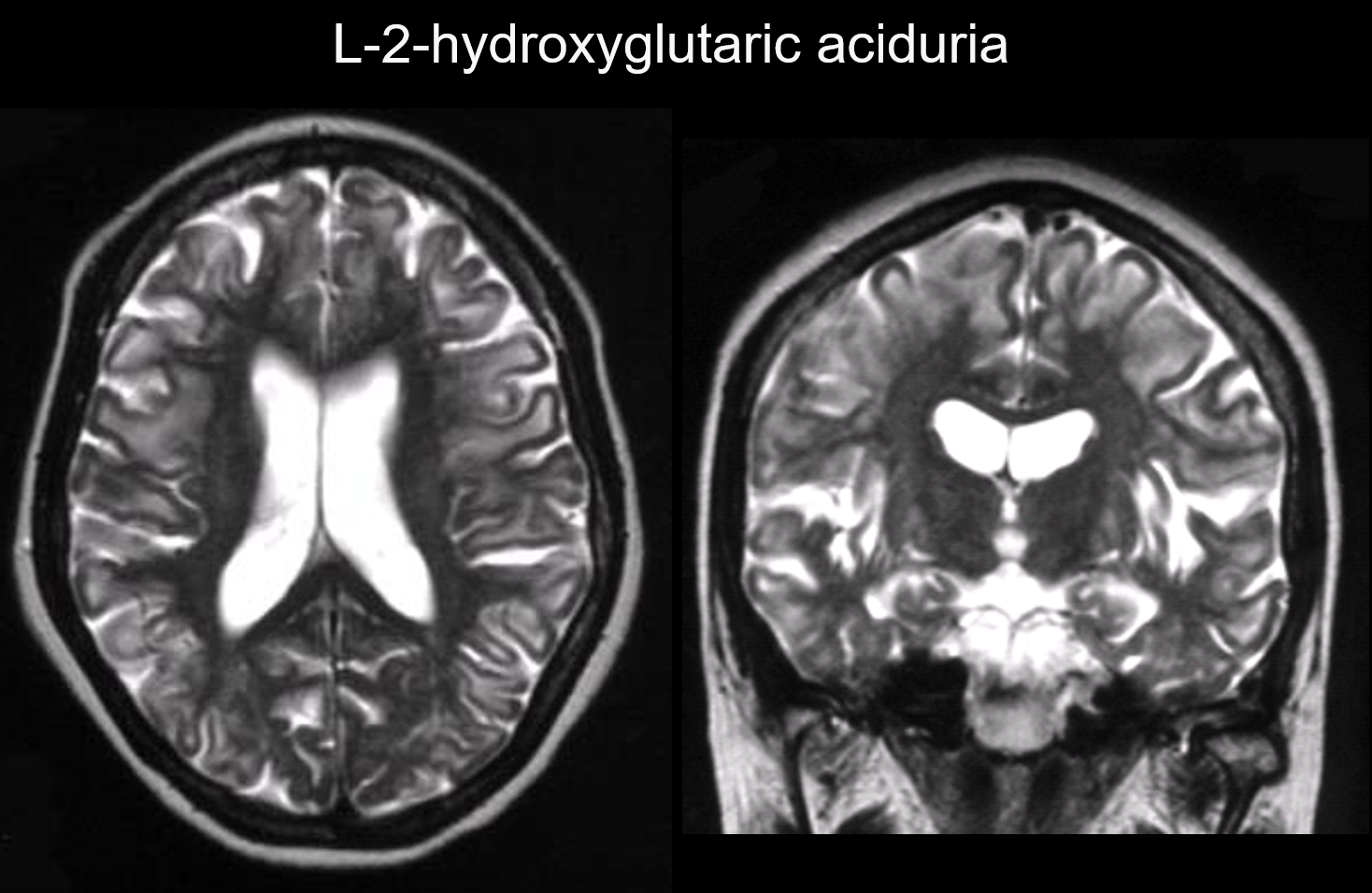
5. Subcorticale predominantie
Bij deze aandoeningen treden laesies voornamelijk op in de subcorticale witte stof, met inbegrip van de U-vezels. Aandoeningen met dit patroon zijn onder meer L-2-hydroxyglutaarzuururie (figuur 6), galactosemie, Kearns-Sayer-syndroom, propionzuuracademie, ureumcyclusstoornissen, en de ziekte van Canavan in een vroeg stadium.
6. Diffuse cerebrale
Bij deze aandoeningen verschijnen abnormale signalen overal in de cerebrale witte stof. Zij vertonen sterke T2-hyperintensiteiten in vergelijking met de T2-signalen die door ongemyeliniseerde witte stof worden geproduceerd (hypomyelinisatie). Naast gevallen van megalencephale leukoencephalopathie met subcorticale cysten en leukoencephalopathie met verdwijnende witte stof, vertonen patiënten met om het even welk type witte stof stoornis uiteindelijk dit patroon naarmate de ziekte vordert.
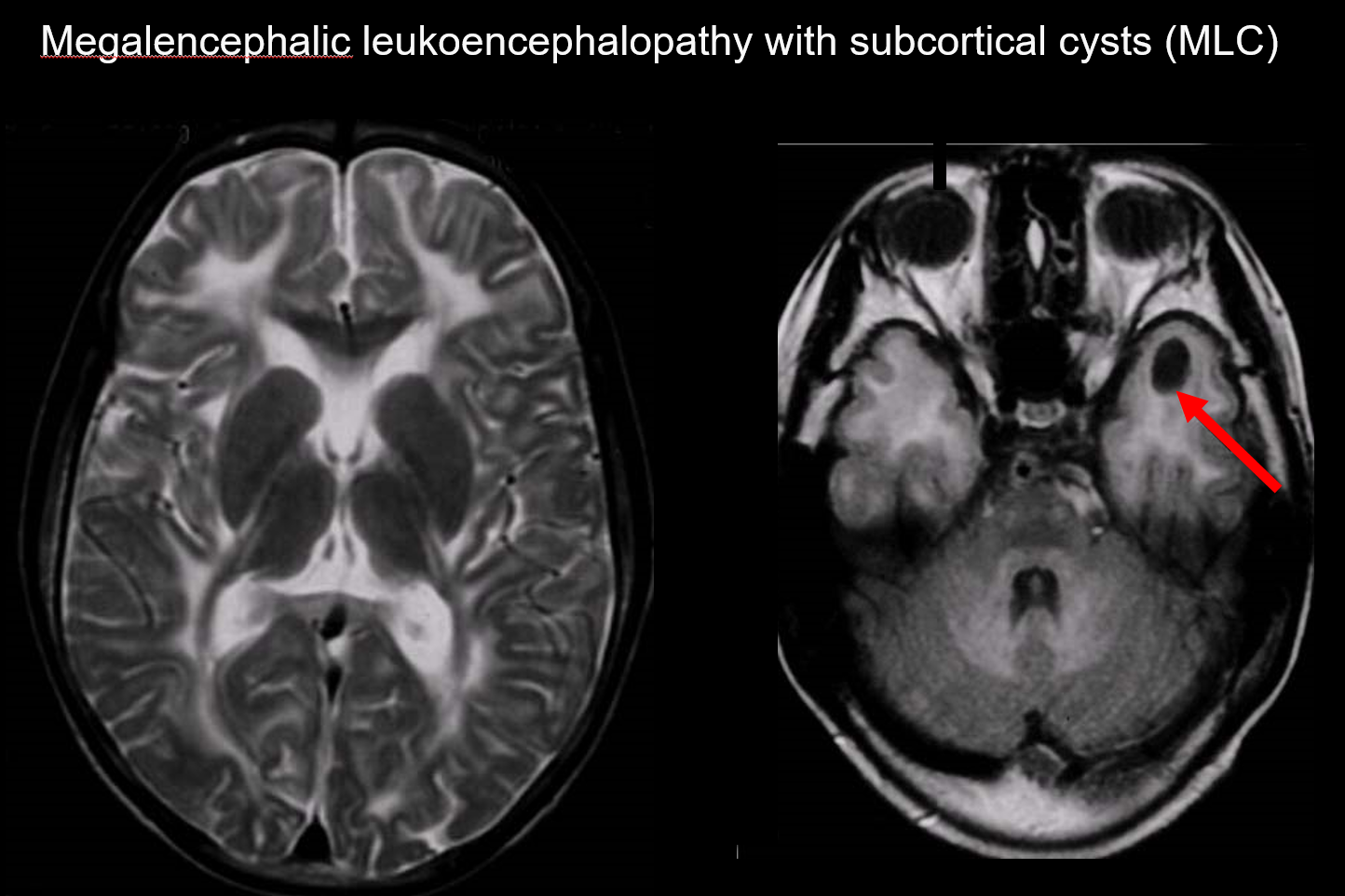
(a) Megalencephale leukoencephalopathie met subcorticale cysten (MLC)
MLC is een autosomaal recessief overervende aandoening die wordt veroorzaakt door een afwijking van het MLC1-gen, en het begin ervan in de kindertijd wordt gekenmerkt door megalocephalie, langzaam voortschrijdende motorische achteruitgang, ataxie, en spasticiteit. MRI onthult karakteristieke wijdverspreide abnormale signalen in de witte stof en milde zwelling van de witte stof, evenals cystevorming in de pariëtale en temporale kwabben (figuur 7).7, 8) T1-gewogen en T2-gewogen beeldvorming onthullen abnormale witte stof, terwijl de cysten allemaal T1 hypointensiteit en T2 hyperintensiteit vertonen, waardoor ze bijzonder moeilijk te detecteren zijn. FLAIR beeldvorming, die cysten (water) visualiseert als hypointensiteiten, is waardevol voor de diagnose. Het komt vaker voor bij Japanners dan vacuolerende megalencephale leukoencephalopathie.
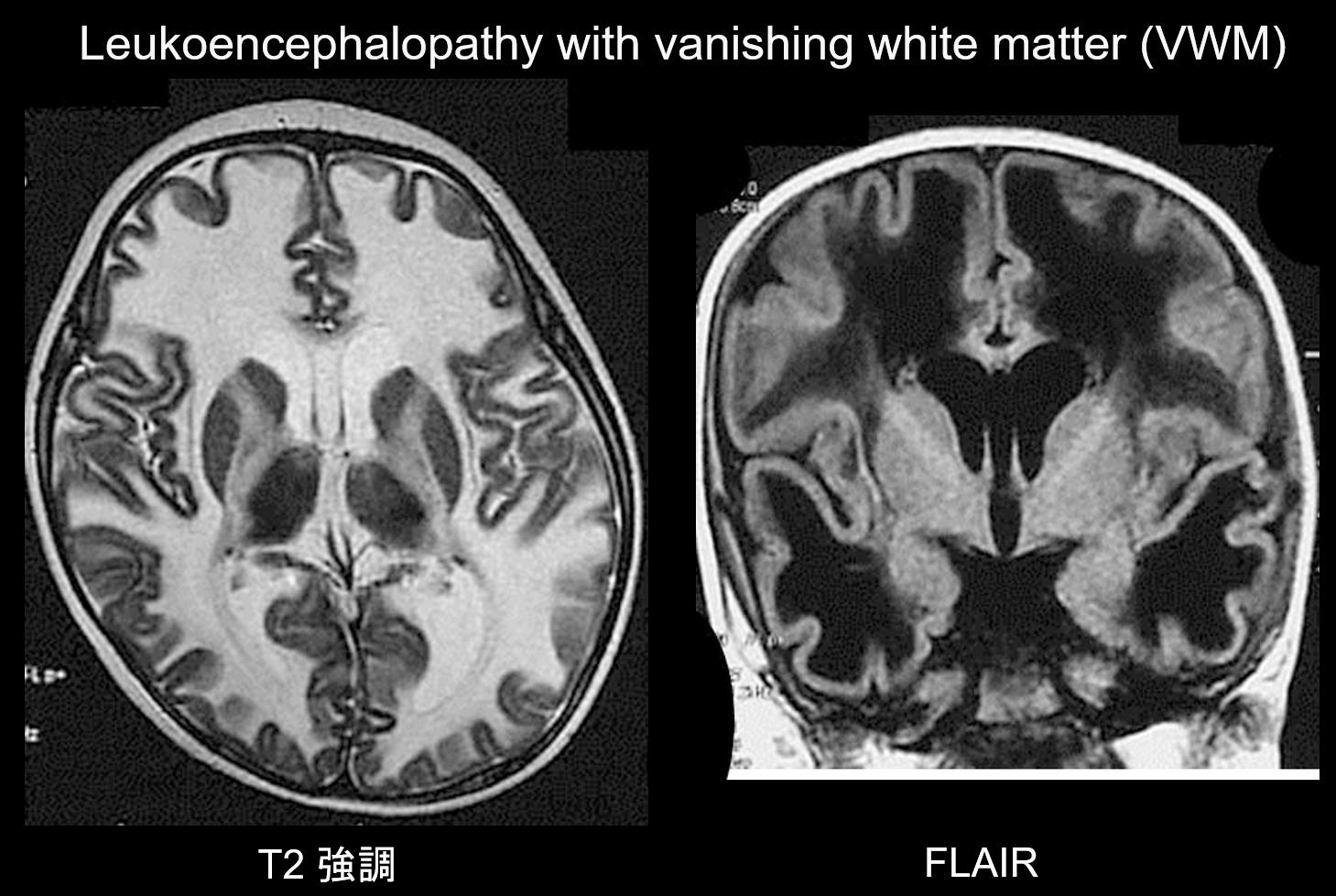
(b) Leukoencephalopathie met verdwijnende witte stof.
Leukoencephalopathie met verdwijnende witte stof (VWM) is een autosomaal recessief overervende aandoening die wordt veroorzaakt door een tekort aan eIF2B, een eiwit dat geassocieerd is met eIF2, dat initiator tRNA overbrengt naar ribosomen. eIF2B bestaat uit vijf verschillende eiwitten, die allemaal verschillende genetische loci hebben. Aangetoond is dat VWM dezelfde aandoening is als cerebellaire ataxie en centrale hypomyelinisatie (CACH) bij kinderen. Patiënten zijn normaal tijdens de neonatale periode en de vroege kinderjaren, maar na de aanvang (meestal op de leeftijd van 2-6 jaar) ontwikkelen zij langzaam progressieve cognitieve regressie, spasticiteit en ataxie. Het is bekend dat deze symptomen verergeren door infectie of klein trauma. De witte hersenstof vertoont wijdverspreide T2 hyperintensiteit en T1 hypointensiteit en wordt geleidelijk vervangen door vloeistof na verloop van tijd (zoals de naam impliceert, de witte stof verdwijnt) (figuur 8). De cystische witte stof bevat bandstructuren waarvan wordt aangenomen dat zij het resterende weefsel vertegenwoordigen. Afwijkende signalen worden ook gezien in de hersenstam, met name in de centrale tegmentale tractus. FLAIR-beeldvorming is waardevol voor de diagnose van deze aandoening.
7. Posterior fossa predominantie of prominentie
Deze aandoeningen worden gekenmerkt door laesies overwegend in de hersenstam en het cerebellum. Cerebellaire witte stof laesies kunnen worden veroorzaakt door aandoeningen waaronder cerebrotendineuze xanthomatose (CTX), peroxisomale aandoeningen, ziekte van Alexander, leukoencefalopathie met betrokkenheid van hersenstam en ruggenmerg en lactaatverhoging (LBSL), maple syrup urine disease, histiocytose, en heroïne- en cocaïnetoxiciteit. Hersenstamlaesies kunnen worden veroorzaakt door aandoeningen zoals de ziekte van Alexander, LSBL, en polyglucosanziekte bij volwassenen. Midden cerebellaire pedunkel laesies worden gezien in fragiele X syndroom en volwassen autosomaal dominante leukodystrofie gerelateerd aan een lamin B1 duplicatie.
8. Multifocale laesies
In tegenstelling tot aandoeningen die de confluente laesies beschreven in 2-7 hierboven produceren, resulteren de aandoeningen in deze sectie in multifocale (verspreide) laesies. Zij omvatten infecties zoals het TORCH-syndroom (door congenitale cytomegalovirusinfectie of een andere oorzaak) en brucellose; ontstekingsaandoeningen zoals acute gedissemineerde encefalomyelitis (ADEM), multiple sclerose (MS), en neuromyelitis optica (NMO); vasculopathieën zoals autosomaal-dominante cerebrale arteriopathie met subcorticale infarcten en leukoencefalopathie (CADASIL), atherosclerose, amyloïde angiopathie, COL4A1-geassocieerde cerebrale small-vessel disease, de ziekte van Fabry en het Susac-syndroom; en erfelijke aandoeningen zoals mitochondriale ziekte, L-2-hydroxyglutaarzuuraurie, mucopolysaccharidose (MPS), en chromosoomafwijkingen (zoals 6p-syndroom).
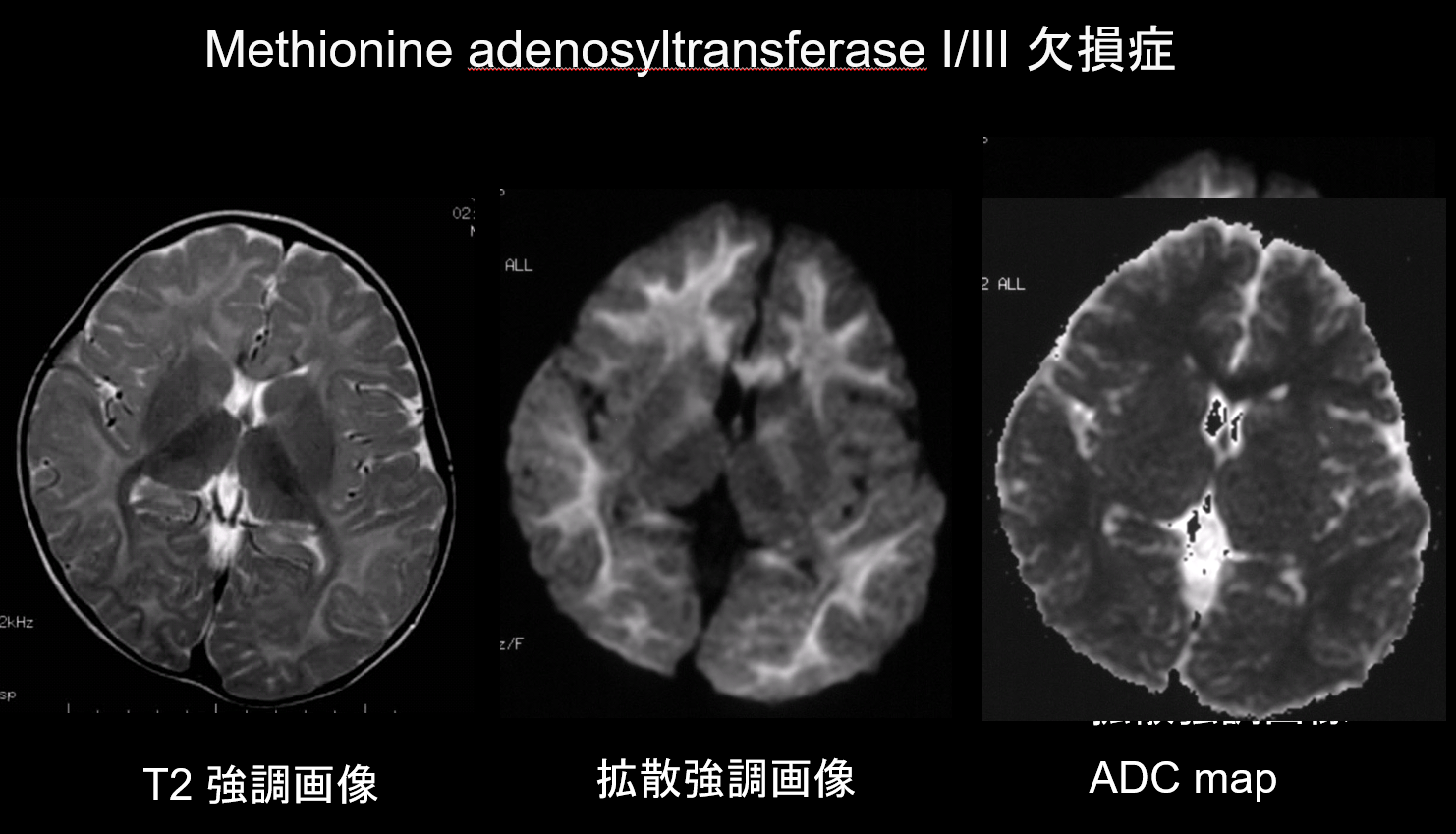
9. Lesies met een lage diffusiecapaciteit
In zowel demyelinisatie als hypomyelinisatie, de belangrijkste pathologieën van witte stof aandoeningen, is er een afname van de hoeveelheid myeline, die diffusie beperkt, en de overeenkomstige toename van extracellulaire vloeistof resulteert in T2 hyperintensiteiten met een hoge schijnbare diffusiecoëfficiënt (ADC). Het is zeldzaam dat witte stof aandoeningen zowel T2 hyperintensiteiten als een lage ADC vertonen, en deze combinatie is dan ook van hoge diagnostische waarde. Aandoeningen die gekenmerkt worden door de aanwezigheid van intramyelinisch oedeem binnen de myelineschede en in de openingen tussen de scheden vertonen een lage ADC. Hiertoe behoren maple syrup urine disease, methionine adenosyltransferase I/III deficiëntie (figuur 9), fenylketonurie, non-ketotic hyperglycinemie, en de ziekte van Canavan. De ziekte van Krabbe en metachromatische leukodystrofie kunnen ook een lage ADC in sommige witte stof laesies vertonen, aangezien intramyelinisch oedeem kan optreden tijdens de acute fase van demyelinisatie.
- Van der Knaap MS, Valk J. Classification of myelin disorders. In Van der Knaap MS, Valk J, eds. Magnetic resonance of myelination and myelin disorders. 3rd ed. Berlin: Springer, 2005, 20-24.
- Schiffmann R, van der Knaap MS. An MRI-based approach to the diagnosis of white matter disorders. Neurology 2009; 72: 750-759
- Takanashi J. Diagnostic imaging of white matter disorders. Journal of the Japan Pediatric Society 2007; 111: 1243-1254.
- Van der Knaap MS, Breiter SN, Naidu S, et al. Defining and categorizing leukoencephalopathies of unknown origin: MR imaging approach. Radiology 1999; 213: 121-133.