Imagerie diagnostique des anomalies de la substance blanche | Hypomyélinisation cérébrale congénitale ; réseau pour la maladie de Pelizaeus-Merzbacher et les troubles associés
Imagerie diagnostique des anomalies de la substance blanche
Junichi TAKANASHI, département de pédiatrie, Université médicale des femmes de Tokyo, Centre médical Yachiyo
Introduction
Dans cet article, je présente l’approche employée pour le diagnostic des troubles qui apparaissent comme des signaux anormaux dans la substance blanche cérébrale sur l’imagerie par résonance magnétique (IRM), de l’imagerie au diagnostic. Les troubles qui affectent principalement la substance blanche sont généralement appelés « leucoencéphalopathie » ou « white matter disorders » en anglais1, 2) 3) Un autre terme, leucodystrophie, est parfois confondu avec la dégénérescence de la substance blanche, mais il s’agit en fait d’un spectre plus étroit de troubles ayant une composante génétique (troubles démyélinisants héréditaires).
Classification des troubles de la substance blanche basée sur l’imagerie
L’avènement de l’IRM a considérablement amélioré notre capacité à détecter les lésions de la substance blanche du système nerveux central. De nombreuses formes connues de troubles de la substance blanche présentent des signes spécifiques à l’IRM, ce qui est utile pour leur diagnostic. L’identification des schémas d’anomalies de la substance blanche observés en IRM (imagerie pondérée en T1, pondérée en T2 ou FLAIR) permet de réduire facilement les possibilités de diagnostics différentiels multiples. La classification de Schiffmann et van der Kamp des troubles de la substance blanche en fonction des résultats de l’IRM présente un intérêt pratique2, 4) (figure 1, tableau 1). Même si elle ne conduit pas à un diagnostic final, la classification des résultats d’imagerie peut conduire à la découverte ultérieure d’un nouveau trouble. Je décris ici les troubles de la substance blanche en fonction des classifications IRM ci-dessus, et j’explique les principaux types de troubles.
Tableau 1. Liste des troubles en fonction des profils IRM
- Dominance frontale
Maladie d’Alexander, variante frontale de l’adrénoleucodystrophie liée au chromosome X (ALD), leucodystrophie métachromatique (MLD), leucodystrophie neuroaxonale avec sphéroïdes. - Dominance pariéto-occipitale
Adrénoleucodystrophie liée à l’X (ALD), maladie de Krabbe,troubles peroxysomaux à début précoce, hypoglycémie néonatale. - Dominance périventriculaire
Leucodystrophie métachromatique (MLD), maladie de Krabbe, syndrome de Sjögren-Larsson, maladie des corps polyglucosan de l’adulte, leucoencéphalopathie avec atteinte du tronc cérébral et de la moelle épinière et élévation des lactates (LBSL), leucomalacie périventriculaire (PVL), encéphalopathie à VIH, lipofuscinoses céroïdes neuronales à début tardif. - Prédominance sous-corticale
Acidurie L-2-hydroxyglutarique, galactosémie, syndrome de Kearns-Sayer, académie propionique, troubles du cycle de l’urée, maladie de Canavan. - Leucoencéphalopathie cérébrale diffuse
avec kystes sous-corticaux (MLC), leucoencéphalopathie avec disparition de la substance blanche (VWM), dystrophie musculaire congénitale déficiente en mérosine, maladie mitochondriale, déficit en cofacteur de molybdène, déficit en sulfite oxydase, cas avancés de troubles de la substance blanche. - Prédominance ou proéminence de la fosse postérieure
Lésions du cervelet et des pédoncules cérébelleux : xanthomatose cérébrotendineuse (CTX), troubles peroxysomaux, maladie d’Alexander, leucoencéphalopathie avec atteinte du tronc cérébral et de la moelle épinière et élévation des lactates (LBSL), maladie urinaire du sirop d’érable, histiocytose, leucodystrophie autosomique dominante de l’adulte liée à une duplication de la lamine B1, toxicité de l’héroïne et de la cocaïne.
Lésions du tronc cérébral : Maladie d’Alexander, LSBL, troubles peroxysomaux, maladie de Wilson, maladie du polyglucosan adulte, syndrome de Leigh, atrophie dentatorubropallidoluysienne (DRPLA), maladie du corps polyglucosan adulte, leucodystrophie adulte autosomique dominante liée à une duplication de la lamelle B1. - Lésions multifocales
Syndrome deTORCH (infection congénitale à cytomégalovirus), brucellose, encéphalomyélite aiguë disséminée (ADEM), sclérose en plaques (SEP), neuromyélite optique (NMO), artériopathie cérébrale autosomique dominante avec infarctus sous-corticaux et leucoencéphalopathie (CADASIL), athérosclérose, angiopathie amyloïde, maladie des petits vaisseaux cérébraux associée au COL4A1, maladie de Fabry, syndrome de Susac, maladie mitochondriale, acidurie L-2-hydroxyglutarique, mucopolysaccharidose (MPS), anomalies chromosomiques (comme le syndrome 6p). - Lésions à faible capacité de diffusion
Maladie des urines du sirop d’érable, déficit en méthionine adénosyltransférase I/III, phénylcétonurie, hyperglycinémie non cétosique, maladie de Canavan, lésions actives dans la maladie de Krabbe et leucodystrophie métachromatique.
1. Hypomyélinisation de la substance blanche cérébrale
Ceci fait référence à un groupe de troubles dans lesquels la formation de la gaine de myéline est altérée ou retardée, et ses images ressemblent à celles des nouveau-nés avec une myélinisation immature. Sur les images pondérées en T2, la substance blanche apparaît de manière caractéristique comme une hyperintensité généralisée qui est faible par rapport au cortex. Pour plus de détails, voir le site Web de l’hypomyélinisation cérébrale congénitale.
Si les lésions de la substance blanche ne sont pas compatibles avec une hypomyélinisation de la substance blanche cérébrale, il faut déterminer si elles sont confluentes ou multiples.2) Les lésions confluentes de la substance blanche sont généralement dues à une dégénérescence héréditaire de la substance blanche (leucodystrophie) et, dans la plupart des cas, sont bilatéralement symétriques. Les lésions multiples de la substance blanche sont généralement asymétriques et acquises. Les lésions confluentes de la substance blanche sont subdivisées en catégories 2 à 7 ci-dessous.
2. Prédominance frontale
Dans ce groupe de troubles, des lésions étendues de la substance blanche sont présentes de manière prédominante dans le lobe frontal. Ils comprennent la maladie d’Alexander, la variante frontale de l’adrénoleucodystrophie liée à l’X (ALD), la leucodystrophie métachromatique (MLD) et la leucodystrophie neuroaxonale avec sphéroïdes.
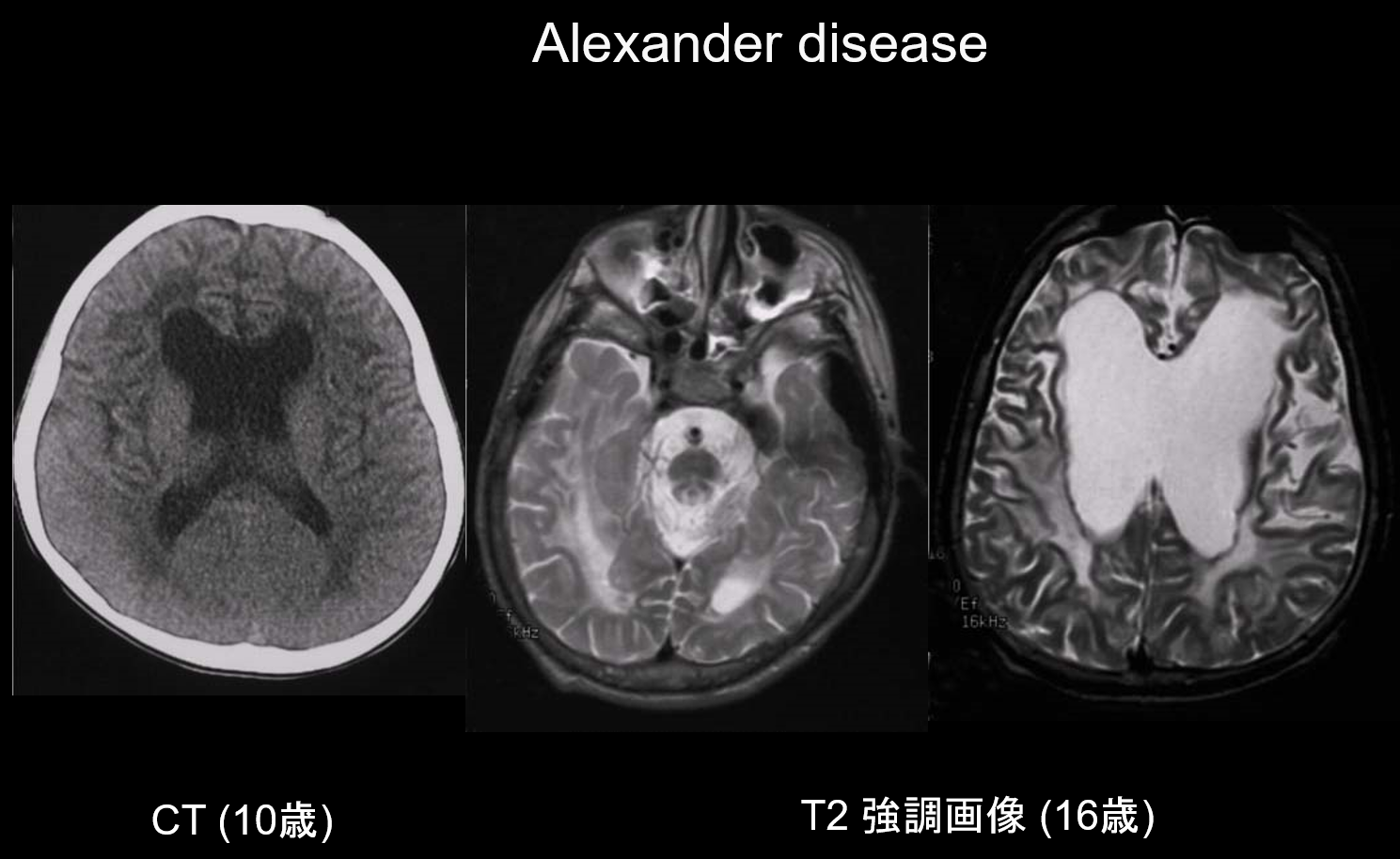
(a) Maladie d’Alexander.
La maladie d’Alexander est une maladie héréditaire autosomique dominante causée par une mutation du gène GFAP sur le chromosome 17q21. Elle se traduit par l’accumulation de fibres de Rosenthal dans les cellules gliales stellaires. Ces fibres sont composées de GFAP et de protéines de stress (αB-cristalline et HSP27). La maladie d’Alexander se manifeste principalement dans la petite enfance, entre l’âge de 3 mois et 2 ans, avec l’apparition d’une mégalencéphalie, d’un retard de développement, d’une paralysie spastique et d’une épilepsie. À l’IRM, elle peut présenter (i) des lésions étendues de la substance blanche, principalement dans le lobe frontal ; (ii) une margination hyperintense en T1 et hypointense en T2 autour des ventricules latéraux ; (iii) des lésions dans les ganglions de la base et le thalamus ; (iv) des lésions du tronc cérébral ; et (v) un renforcement du contraste des lésions actives (figure 2). Dans les premiers stades, avec les lésions de la substance blanche et du putamen, on observe un gonflement qui peut progressivement entraîner une atrophie ou la formation de kystes.
3. Prédominance pariéto-occipitale
La principale caractéristique de ce groupe de troubles est la présence de lésions de la substance blanche pariéto-occipitale. Ils comprennent l’adrénoleucodystrophie liée à l’X (ALD), la maladie de Krabbe, les troubles peroxysomaux à début précoce et l’hypoglycémie néonatale.
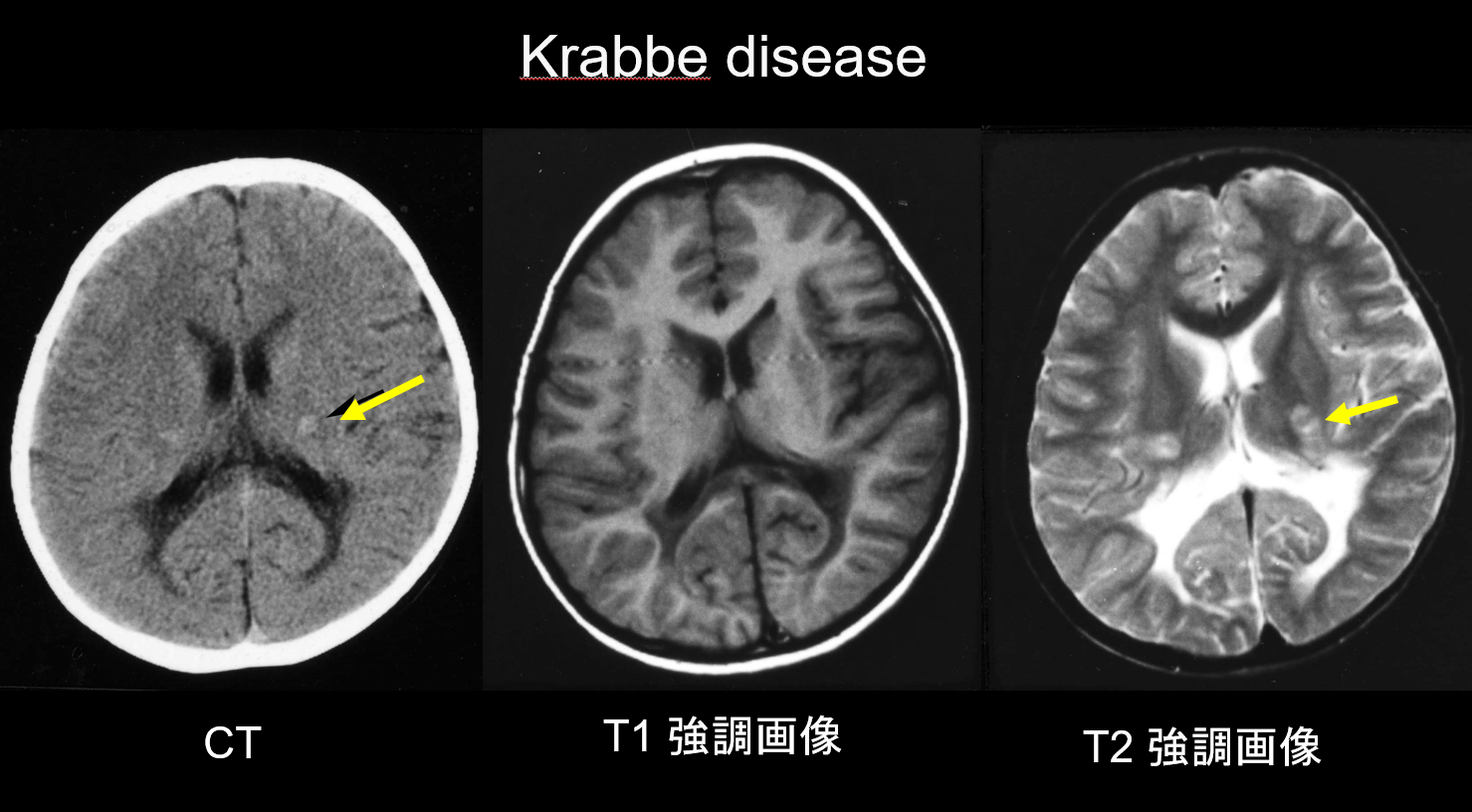
(a) Maladie de Krabbe.
La maladie de Krabbe est une maladie héréditaire autosomique récessive (maladie de stockage lysosomal) causée par un déficit en galactosylcéramidase (chromosome 14q31), dans laquelle l’accumulation de psychosine hautement cytotoxique serait à l’origine d’une démyélinisation généralisée. De grandes cellules multinucléées appelées « cellules globoïdes » apparaissent également. En fonction de l’âge auquel elle apparaît, la maladie est classée comme infantile, infantile à déclenchement tardif, juvénile ou adulte. La plupart des cas sont infantiles et commencent par l’apparition de fièvre, d’irritabilité, de difficultés d’alimentation, de retard de développement, de neuropathie périphérique, de spasticité et d’atrophie du nerf optique à l’âge de 3 à 6 mois. Au cours des premiers stades, la tomodensitométrie (TDM) révèle une hyperdensité caractéristique dans le thalamus et la corona radiata. On pense que cela reflète des cellules globoïdes de haute densité et une prolifération gliale. L’IRM peut également montrer une hyperintensité en T1 et une hypointensité en T2 autour des ventricules, ainsi que des structures linéaires similaires à celles observées dans la MLD (figure 3). Le noyau denté cérébelleux, la substance blanche cérébelleuse et le tractus pyramidal du tronc cérébral présentent une hyperintensité T2 dès un stade précoce.
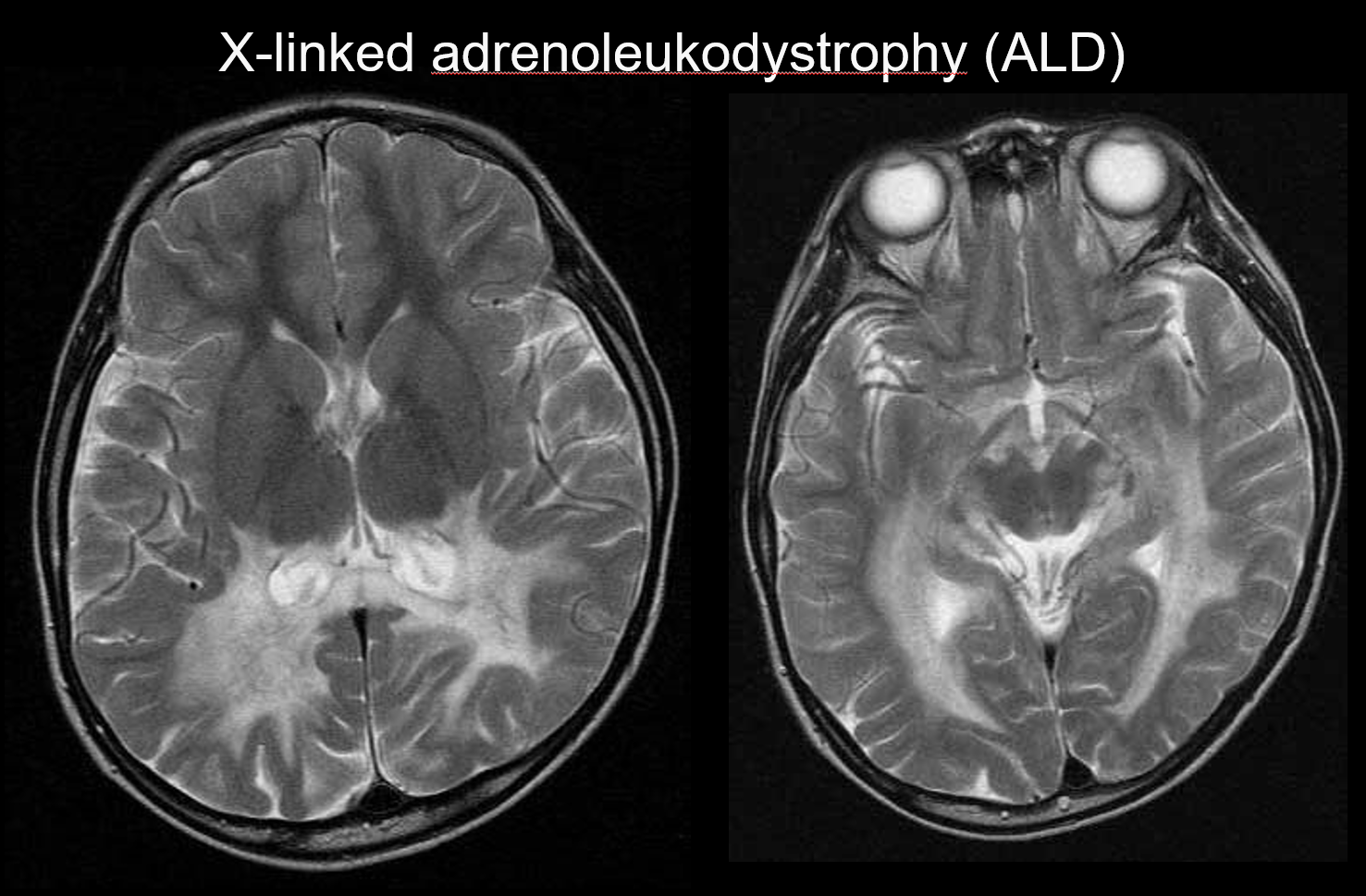
(b) Adrénoleucodystrophie liée à l’X
L’adrénoleucodystrophie liée à l’X (ALD) est une maladie héréditaire récessive liée à l’X (trouble peroxysomal) causée par une anomalie du gène ABCD1 (chromosome Xq28). L’altération de la β-oxydation entraîne l’accumulation d’acides gras à très longue chaîne dans la substance blanche cérébrale et les glandes surrénales, provoquant une démyélinisation et une insuffisance surrénale. L’ALD est classée en trois catégories : les formes cérébrales de l’enfant, de l’adolescent et de l’adulte, l’adrénomyéloneuropathie (AMN) et la maladie d’Addison uniquement. La forme cérébrale de l’enfant se développe à l’âge de 5 à 8 ans, avec l’apparition de symptômes tels qu’une détérioration intellectuelle, une démarche spastique et des troubles de la vision et de l’audition. Sur le plan pathologique, la démyélinisation progresse depuis la substance blanche entourant le trigone du ventricule latéral jusqu’au splénium du corps calleux, s’étendant progressivement sur le plan antérolatéral. Reflétant la pathologie de la maladie, des hyperintensifications T2 et des hypointensifications T1 symétriques s’étendant antérolatéralement à partir de la substance blanche entourant le trigone du ventricule latéral sont apparentes à l’IRM, avec un rehaussement de contraste évident sur les bords (Figure 4). Des lésions du tractus corticospinal sont également évidentes.
4. Prédominance périventriculaire
Ces troubles sont principalement caractérisés par des lésions de la substance blanche entourant les ventricules latéraux, la substance blanche sous-corticale (fibres U) étant préservée. Ce schéma est observé dans de nombreux troubles, y compris le MLD, et est donc comparativement non spécifique. Des signaux légèrement anormaux autour des ventricules latéraux sont également observés dans la dégénérescence corticale, en particulier les lipofuscinoses céroïdes neuronales qui se développent après la naissance.
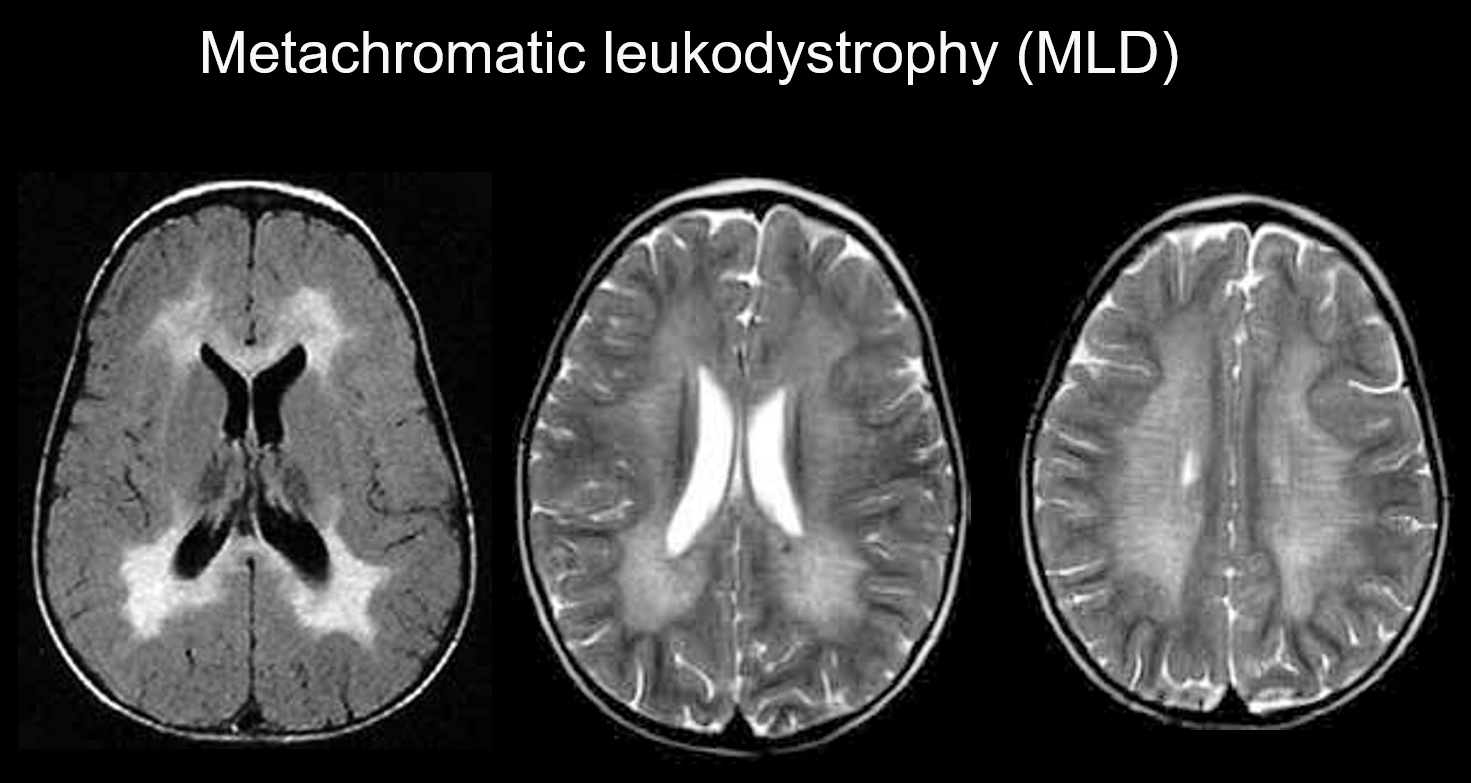
(a) Leucodystrophie métachromatique.
La leucodystrophie métachromatique est une maladie héréditaire autosomique récessive (trouble du stockage lysosomal) due à un déficit en arylsulfatase-A (chromosome 22q13.31), dans laquelle l’accumulation de sulfatide hautement toxique entraîne une démyélinisation. Selon l’âge auquel elle apparaît, elle est classée comme congénitale, infantile, juvénile ou adulte. Ses symptômes comprennent une régression cognitive, une paralysie spastique, des mouvements involontaires, une neuropathie périphérique et une atrophie du nerf optique. Elle se manifeste sur l’imagerie pondérée en T2 par des hyperintensités de la substance blanche, principalement autour des ventricules latéraux, et sur l’imagerie pondérée en T1 par de légères hypointensifications. Les lésions ont tendance à se situer principalement dans le lobe frontal. Des bandes d’intensité normale (bandes tigrées) peuvent être évidentes au sein des signaux anormaux généralisés dans la substance blanche (figure 5). Ceux-ci seraient dus à la préservation partielle de la gaine de myéline dans l’espace périvasculaire et à l’accumulation de produits de dégradation de la gaine de myéline dans les macrophages.
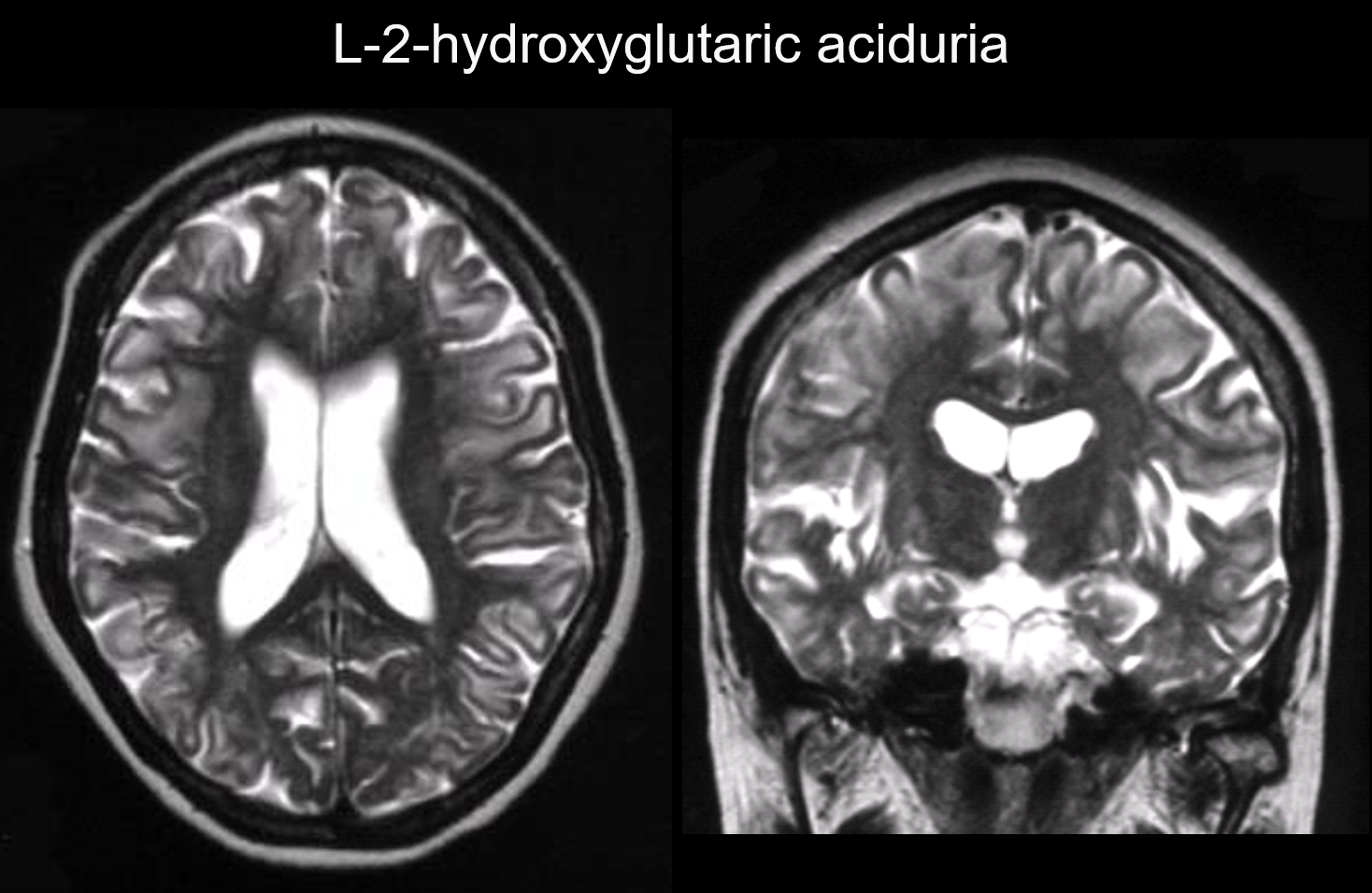
5. Prédominance sous-corticale
Dans ces troubles, les lésions se produisent principalement dans la substance blanche sous-corticale, y compris les fibres U. Les troubles présentant ce schéma incluent l’acidurie L-2-hydroxyglutarique (figure 6), la galactosémie, le syndrome de Kearns-Sayer, l’académie propionique, les troubles du cycle de l’urée et la maladie de Canavan à un stade précoce.
6. Cérébral diffus
Dans ces troubles, des signaux anormaux apparaissent dans toute la substance blanche cérébrale. Ils présentent de fortes hyperintensifications T2 par rapport aux signaux T2 produits par la substance blanche non myélinisée (hypomyélinisation). Outre les cas de leucoencéphalopathie mégalencéphalique avec kystes sous-corticaux et de leucoencéphalopathie avec disparition de la substance blanche, les patients atteints de tout type de trouble de la substance blanche finissent par présenter ce schéma au fur et à mesure que la maladie progresse.
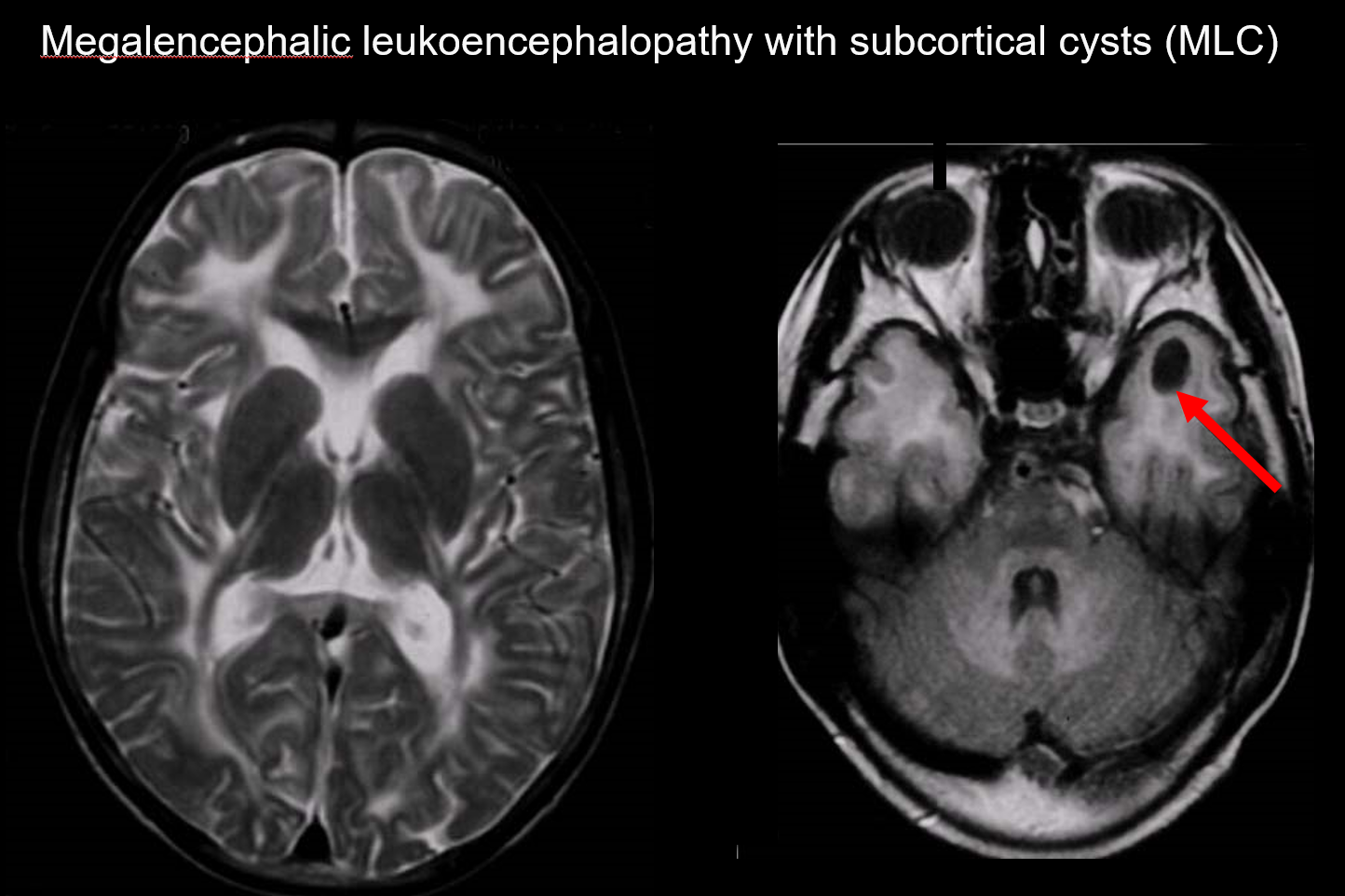
(a) Leucoencéphalopathie mégalencéphalique avec kystes sous-corticaux (MLC)
La MLC est une maladie héréditaire autosomique récessive causée par une anomalie du gène MLC1, et son apparition dans la petite enfance est marquée par une mégalocéphalie, une détérioration motrice à progression lente, une ataxie et une spasticité. L’IRM révèle des signaux anormaux généralisés caractéristiques dans la substance blanche et un léger gonflement de celle-ci, ainsi que la formation de kystes dans les lobes pariétal et temporal (Figure 7).7, 8) L’imagerie pondérée en T1 et en T2 révèle une substance blanche anormale, tandis que les kystes présentent tous une hypointensité en T1 et une hyperintensité en T2, ce qui les rend particulièrement difficiles à détecter. L’imagerie FLAIR, qui visualise les kystes (eau) sous forme d’hypointensifications, est précieuse pour son diagnostic. Elle est plus fréquente chez les Japonais que la leucoencéphalopathie mégalencéphalique vacuolante.
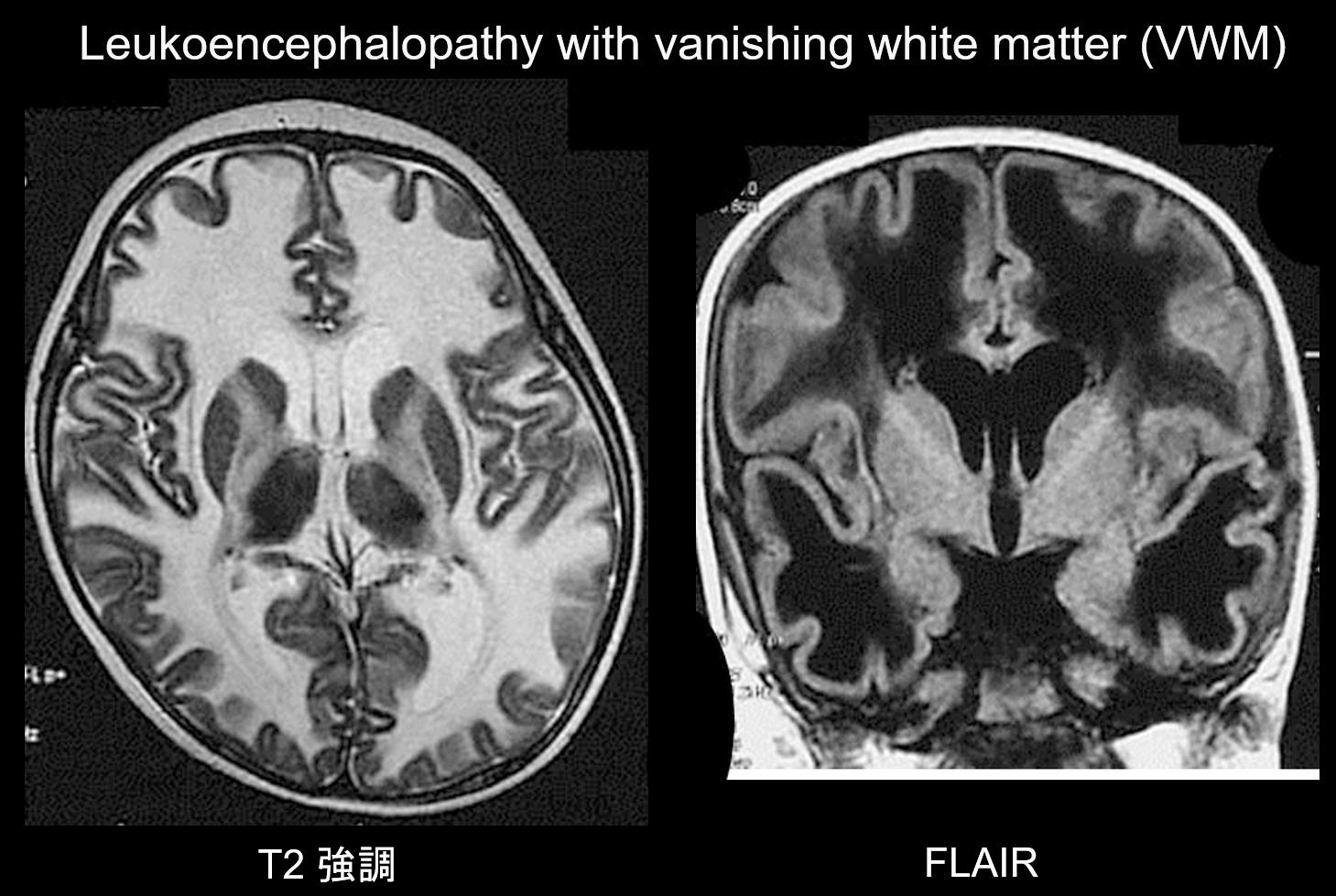
(b) Leucoencéphalopathie avec disparition de la substance blanche.
La leucoencéphalopathie avec substance blanche évanescente (VWM) est une maladie héréditaire autosomique récessive causée par une déficience de eIF2B, une protéine associée à eIF2, qui transfère l’ARNt initiateur aux ribosomes. eIF2B se compose de cinq protéines différentes, qui ont toutes des loci génétiques différents. Il a été démontré que le VWM est le même trouble que l’ataxie cérébelleuse infantile et l’hypomyélinisation centrale (CACH). Les patients sont normaux pendant la période néonatale et la petite enfance, mais après le début de la maladie (généralement entre 2 et 6 ans), ils développent une régression cognitive lentement progressive, une spasticité et une ataxie. On sait que ces symptômes sont exacerbés par une infection ou un traumatisme mineur. La substance blanche cérébrale présente une hyperintensité T2 et une hypointensité T1 généralisées et est progressivement remplacée par du liquide au fil du temps (comme son nom l’indique, la substance blanche disparaît) (figure 8). La substance blanche kystique contient des structures en bandes qui sont censées représenter le tissu restant. Des signaux anormaux sont également observés dans le tronc cérébral, en particulier dans le tractus tegmental central. L’imagerie FLAIR est précieuse pour le diagnostic de ce trouble.
7. Prédominance ou proéminence de la fosse postérieure
Ces troubles sont caractérisés par des lésions prédominant dans le tronc cérébral et le cervelet. Les lésions de la substance blanche cérébelleuse peuvent être causées par des troubles tels que la xanthomatose cérébrotendineuse (CTX), les troubles peroxysomaux, la maladie d’Alexander, la leucoencéphalopathie avec atteinte du tronc cérébral et de la moelle épinière et élévation des lactates (LBSL), la maladie urinaire du sirop d’érable, l’histiocytose et la toxicité de l’héroïne et de la cocaïne. Les lésions du tronc cérébral peuvent être causées par des troubles tels que la maladie d’Alexander, la LBSL et la maladie du polyglucosan de l’adulte. Des lésions du pédoncule cérébelleux moyen sont observées dans le syndrome du X fragile et la leucodystrophie autosomique dominante de l’adulte liée à une duplication de la lamine B1.
8. Lésions multifocales
À la différence des troubles qui produisent les lésions confluentes décrites en 2-7 ci-dessus, les troubles de cette section entraînent des lésions multifocales (dispersées). Ils comprennent des infections telles que le syndrome TORCH (dû à une infection congénitale à cytomégalovirus ou à une autre cause) et la brucellose ; des troubles inflammatoires tels que l’encéphalomyélite aiguë disséminée (ADEM), la sclérose en plaques (SEP) et la neuromyélite optique (NMO) ; les vasculopathies telles que l’artériopathie cérébrale autosomique-dominante avec infarctus sous-corticaux et leucoencéphalopathie (CADASIL), l’athérosclérose, l’angiopathie amyloïde, la maladie des petits vaisseaux cérébraux associée au COL4A1, la maladie de Fabry et le syndrome de Susac ; et les affections héréditaires telles que la maladie mitochondriale, l’acidurie L-2-hydroxyglutarique, la mucopolysaccharidose (MPS) et les anomalies chromosomiques (telles que le syndrome 6p).
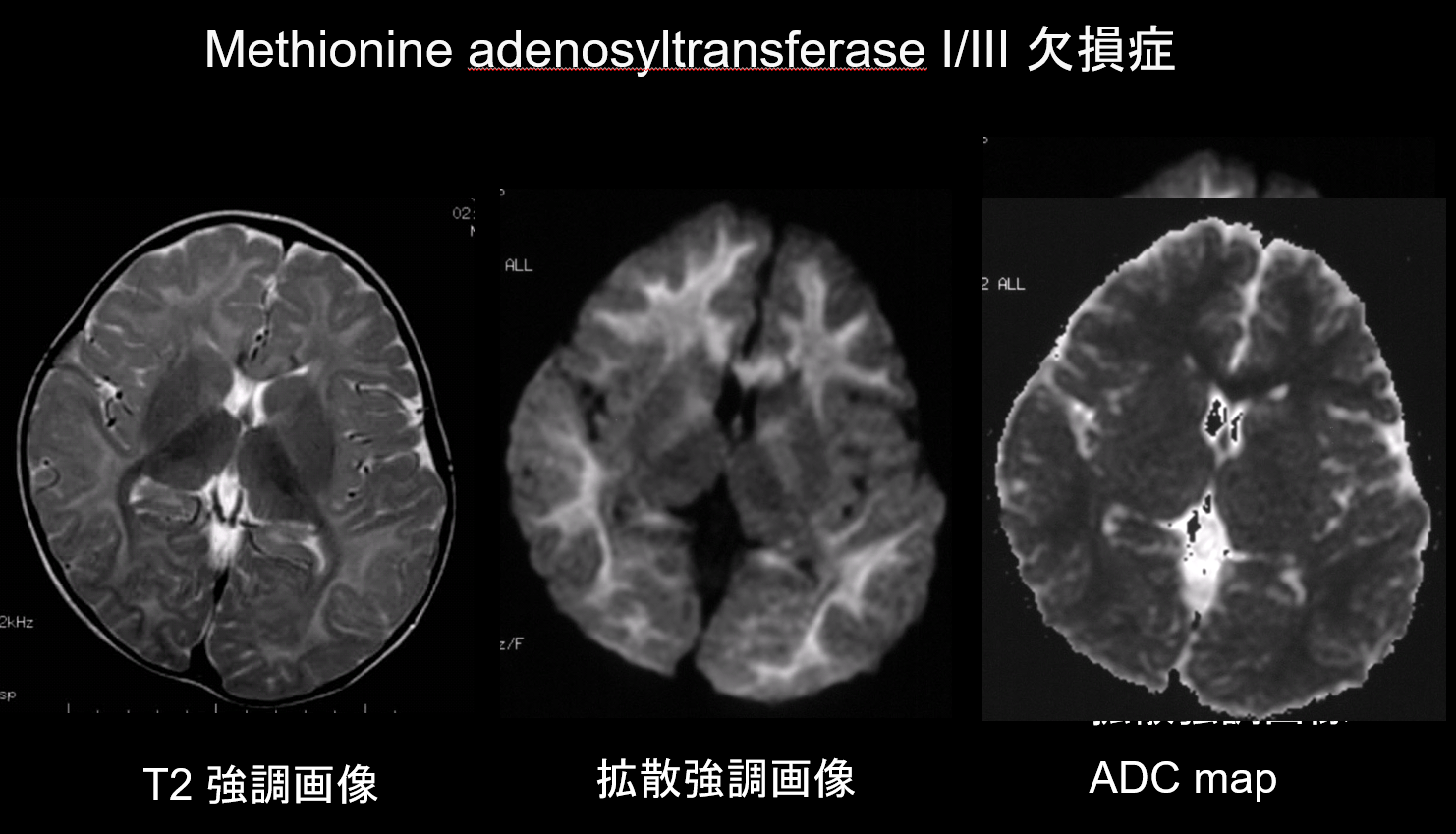
9. Lésions à faible capacité de diffusion
Dans la démyélinisation et l’hypomyélinisation, les principales pathologies des troubles de la substance blanche, il y a une diminution de la quantité de myéline, ce qui limite la diffusion, et l’augmentation correspondante du liquide extracellulaire entraîne des hyperintensifications T2 avec un coefficient de diffusion apparent (ADC) élevé. Il est rare que les troubles de la substance blanche présentent à la fois des hyperintensités T2 et un faible ADC, et cette combinaison a donc une grande valeur diagnostique. Les troubles caractérisés par la présence d’un œdème intramyélinique dans la gaine de myéline et dans les espaces entre les gaines présentent un faible ADC. Il s’agit notamment de la maladie urinaire du sirop d’érable, du déficit en méthionine adénosyltransférase I/III (figure 9), de la phénylcétonurie, de l’hyperglycinémie non cétosique et de la maladie de Canavan. La maladie de Krabbe et la leucodystrophie métachromatique peuvent également présenter un faible ADC dans certaines lésions de la substance blanche, car un œdème intramyélinique peut se produire pendant la phase aiguë de démyélinisation.
- Van der Knaap MS, Valk J. Classification of myelin disorders. In Van der Knaap MS, Valk J, eds. Résonance magnétique de la myélinisation et des troubles de la myéline. 3rd ed. Berlin : Springer, 2005, 20-24.
- Schiffmann R, van der Knaap MS. Une approche basée sur l’IRM pour le diagnostic des troubles de la substance blanche. Neurologie 2009 ; 72 : 750-759
- Takanashi J. Imagerie diagnostique des troubles de la substance blanche. Journal de la société japonaise de pédiatrie 2007 ; 111 : 1243-1254.
- Van der Knaap MS, Breiter SN, Naidu S, et al. Définir et catégoriser les leucoencéphalopathies d’origine inconnue : Approche de l’imagerie par RM. Radiology 1999 ; 213 : 121-133.
.