Diagnóstico por imagen de las anomalías de la sustancia blanca | Hipomielinización cerebral congénita; red para la enfermedad de Pelizaeus-Merzbacher y trastornos relacionados
Diagnóstico por imagen de las anomalías de la sustancia blanca
Junichi TAKANASHI, Departamento de Pediatría, Universidad Médica Femenina de Tokio, Yachiyo Medical Center
Introducción
En este artículo, describo el enfoque empleado para el diagnóstico de los trastornos que aparecen como señales anormales en la materia blanca cerebral en las imágenes de resonancia magnética (IRM), desde las imágenes hasta el diagnóstico. Los trastornos que afectan principalmente a la sustancia blanca suelen denominarse «leucoencefalopatía» o «trastornos de la sustancia blanca» en inglés1, 2) 3) Otro término, leucodistrofia, se confunde a veces con degeneración de la sustancia blanca, pero en realidad se refiere a un espectro más estrecho de trastornos con un componente genético (trastornos desmielinizantes heredados).
Clasificación basada en imágenes de los trastornos de la sustancia blanca
La llegada de la IRM ha mejorado drásticamente nuestra capacidad para detectar lesiones en la sustancia blanca del sistema nervioso central. Muchas formas conocidas de trastornos de la sustancia blanca presentan signos específicos en la RM, lo que resulta útil para su diagnóstico. La identificación de los patrones de anormalidad de la sustancia blanca que se observan en la RM (imágenes ponderadas en T1, T2 o FLAIR) facilita la reducción de las posibilidades en los múltiples diagnósticos diferenciales. La clasificación de Schiffmann y van der Kamp de los trastornos de la sustancia blanca según los hallazgos de la RM es de gran valor práctico2, 4) (Figura 1, Tabla 1). Incluso si esto no conduce a un diagnóstico final, la clasificación de los hallazgos de imagen puede conducir al descubrimiento posterior de un nuevo trastorno. A continuación, describo los trastornos de la sustancia blanca en términos de las clasificaciones de IRM mencionadas, y explico los principales tipos de trastornos.
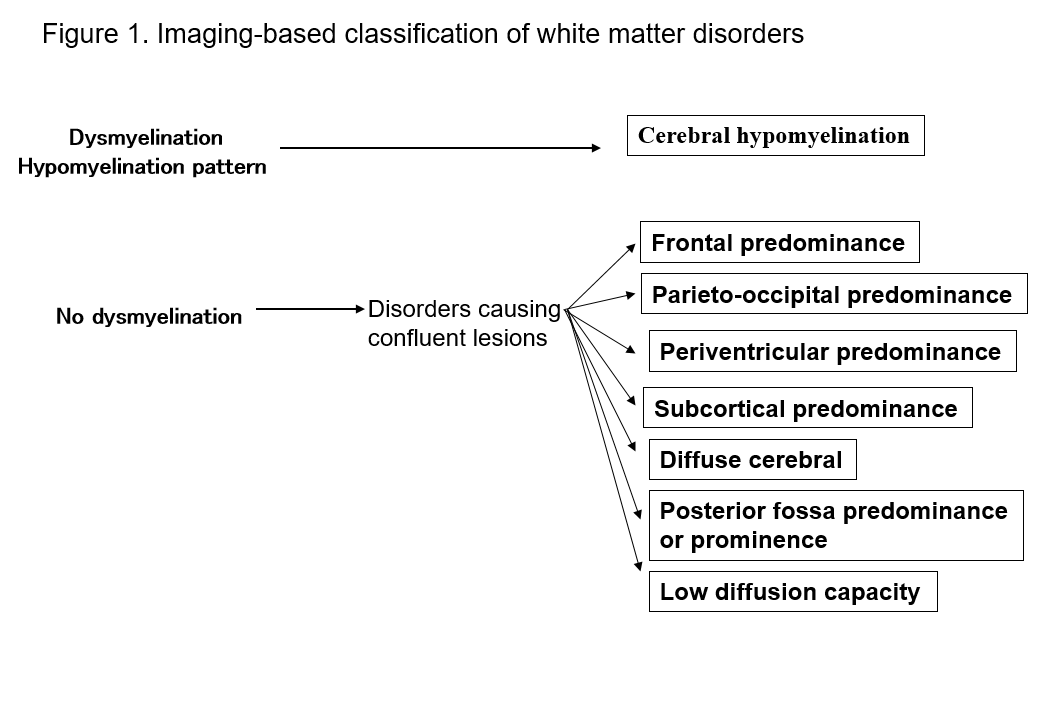
Tabla 1. Lista de trastornos por patrones de RM
- Predominio frontal
Enfermedad de Alexander, variante frontal de la adrenoleucodistrofia ligada al X (ALD), leucodistrofia metacromática (MLD), leucodistrofia neuroaxonal con esferoides. - Predominio parietal-occipital
Adrenoleucodistrofia ligada a X (ALD), enfermedad de Krabbe, trastornos peroxisomales de aparición temprana, hipoglucemia neonatal. - Predominio periventricular
Leucodistrofia metacromática (MLD), enfermedad de Krabbe, síndrome de Sjögren-Larsson, enfermedad del cuerpo poliglucósico del adulto, leucoencefalopatía con afectación del tronco cerebral y la médula espinal y elevación del lactato (LBSL), leucomalacia periventricular (PVL), encefalopatía por VIH, lipofuscinosis ceroidea neuronal de inicio tardío. - Predominio subcortical
Aciduria L-2-hidroxiglutárica, galactosemia, síndrome de Kearns-Sayer, academia propiónica, trastornos del ciclo de la urea, enfermedad de Canavan. - Leucoencefalopatía cerebral difusa con quistes subcorticales (MLC), leucoencefalopatía con desaparición de sustancia blanca (VWM), distrofia muscular congénita por deficiencia de merosina, enfermedad mitocondrial, deficiencia de cofactor de molibdeno, deficiencia de sulfito oxidasa, casos avanzados de trastornos de la sustancia blanca.
- Predominio o prominencia de la fosa posterior
Las lesiones del cerebelo y de los pedúnculos cerebelosos: xantomatosis cerebrotendinosa (CTX), trastornos peroxisomales, enfermedad de Alexander, leucoencefalopatía con afectación del tronco cerebral y de la médula espinal y elevación del lactato (LBSL), enfermedad de la orina del jarabe de arce, histiocitosis, leucodistrofia autosómica dominante del adulto relacionada con una duplicación de la lámina B1, toxicidad por heroína y cocaína.
Las lesiones del tronco cerebral: Enfermedad de Alexander, LSBL, trastornos peroxisomales, enfermedad de Wilson, enfermedad de poliglucosa del adulto, síndrome de Leigh, atrofia dentatorubropallidoluysiana (DRPLA), enfermedad del cuerpo de poliglucosa del adulto, leucodistrofia autosómica dominante del adulto relacionada con una duplicación de la lámina B1. - Lesiones multifocales
Síndrome de Torch (infección congénita por citomegalovirus), brucelosis, encefalomielitis aguda diseminada (ADEM), esclerosis múltiple (EM), neuromielitis óptica (NMO), arteriopatía cerebral autosómica dominante con infartos subcorticales y leucoencefalopatía (CADASIL), aterosclerosis, angiopatía amiloide, enfermedad cerebral de pequeños vasos asociada a COL4A1, enfermedad de Fabry, síndrome de Susac, enfermedad mitocondrial, aciduria L-2-hidroxiglutárica, mucopolisacaridosis (MPS), anomalías cromosómicas (como el síndrome 6p). - Las lesiones con baja capacidad de difusión
Enfermedad de la orina de jarabe de arce, deficiencia de metionina adenosiltransferasa I/III, fenilcetonuria, hiperglicinemia no cetósica, enfermedad de Canavan, lesiones activas en la enfermedad de Krabbe y leucodistrofia metacromática.
1. Hipomielinización de la sustancia blanca cerebral
Se refiere a un grupo de trastornos en los que la formación de la vaina de mielina está alterada o retrasada, y sus imágenes se asemejan a las de los recién nacidos con mielinización inmadura. En las imágenes ponderadas en T2, la sustancia blanca aparece característicamente como una hiperintensidad generalizada que es débil en comparación con la corteza. Para más detalles, véase la página web de la hipomielinización cerebral congénita.
Si las lesiones de la sustancia blanca no son compatibles con la hipomielinización cerebral de la sustancia blanca, debe determinarse si son confluentes o múltiples.2) Las lesiones confluentes de la sustancia blanca suelen deberse a una degeneración hereditaria de la sustancia blanca (leucodistrofia) y en la mayoría de los casos son simétricas bilaterales. Las lesiones múltiples de la sustancia blanca suelen ser asimétricas y adquiridas. Las lesiones confluentes de la sustancia blanca se subdividen a continuación en las categorías 2-7.
2. Predominio frontal
En este grupo de trastornos, las lesiones extensas de la sustancia blanca están presentes predominantemente en el lóbulo frontal. Incluyen la enfermedad de Alexander, la variante frontal de la adrenoleucodistrofia ligada al cromosoma X (ALD), la leucodistrofia metacromática (MLD) y la leucodistrofia neuroaxonal con esferoides.
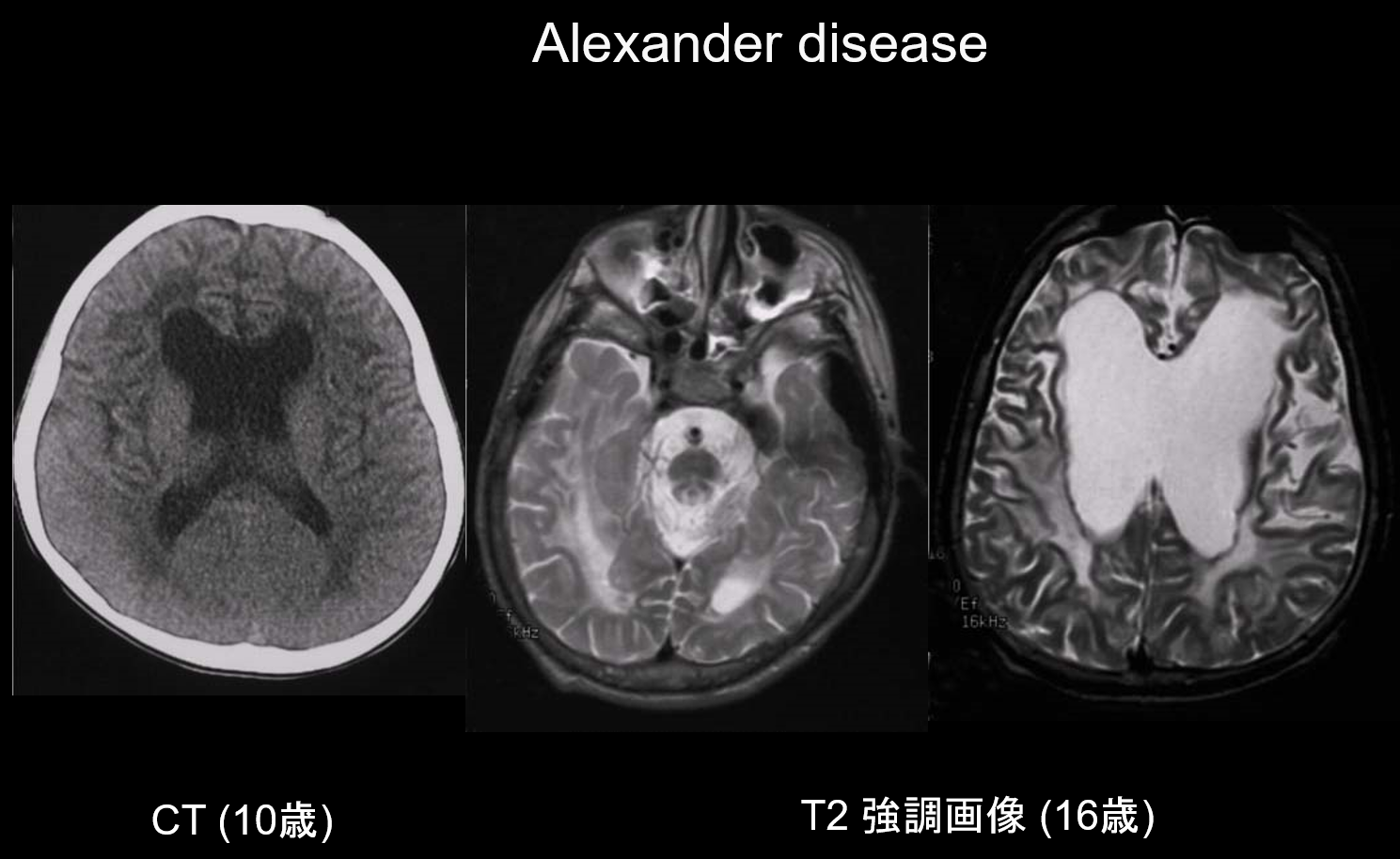
(a) Enfermedad de Alexander.
La enfermedad de Alexander es un trastorno hereditario autosómico dominante causado por una mutación en el gen GFAP del cromosoma 17q21. Da lugar a la acumulación de fibras de Rosenthal en las células gliales estrelladas. Estas fibras están compuestas por GFAP y proteínas de estrés (αB-cristalina y HSP27). La enfermedad de Alexander se presenta principalmente en la infancia, entre los 3 meses y los 2 años, con la aparición de megalencefalia, retraso en el desarrollo, parálisis espástica y epilepsia. En la resonancia magnética, puede mostrar (i) lesiones generalizadas en la sustancia blanca, predominantemente en el lóbulo frontal; (ii) marginación hiperintensa en T1 e hipointensa en T2 alrededor de los ventrículos laterales; (iii) lesiones en los ganglios basales y el tálamo; (iv) lesiones en el tronco encefálico; y (v) realce del contraste de las lesiones activas (Figura 2). En los primeros estadios, junto con las lesiones de la sustancia blanca y del putamen, se observa una inflamación que puede provocar gradualmente la atrofia o la formación de quistes.
3. Predominio parieto-occipital
La característica principal de este grupo de trastornos son las lesiones de la sustancia blanca parieto-occipital. Incluyen la adrenoleucodistrofia (ALD) ligada al cromosoma X, la enfermedad de Krabbe, los trastornos peroxisomales de aparición temprana y la hipoglucemia neonatal.
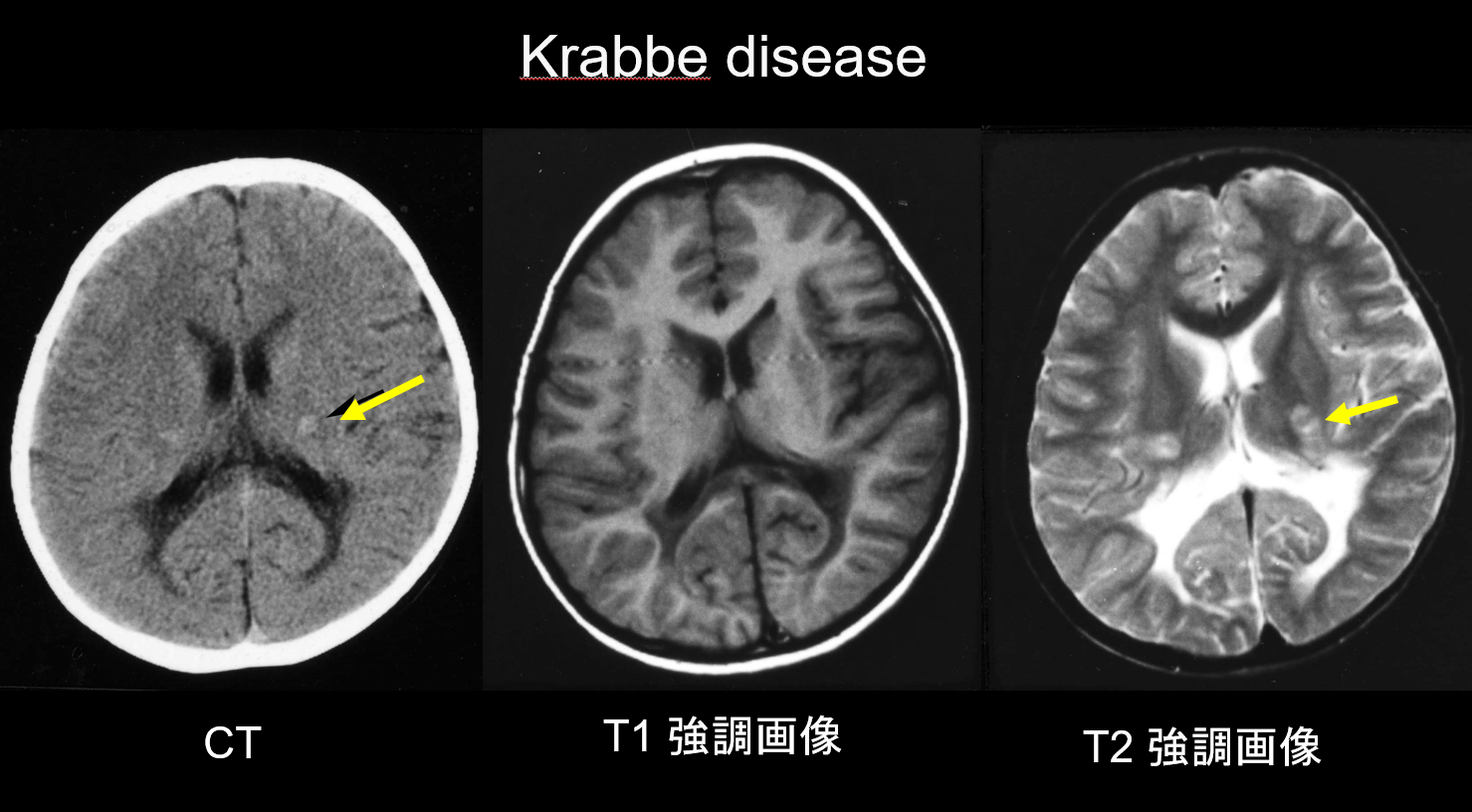
(a) Enfermedad de Krabbe.
La enfermedad de Krabbe es un trastorno hereditario autosómico recesivo (enfermedad de almacenamiento lisosómico) causado por la deficiencia de galactosilceramidasa (cromosoma 14q31), en el que se cree que la acumulación de psicosina altamente citotóxica provoca una desmielinización generalizada. También aparecen células grandes y multinucleadas llamadas «células globoides». Dependiendo de la edad en la que aparece, se clasifica como enfermedad infantil, infantil de inicio tardío, de inicio juvenil o de inicio adulto. La mayoría de los casos son infantiles y comienzan con la aparición de fiebre, irritabilidad, dificultad para alimentarse, retraso en el desarrollo, neuropatía periférica, espasticidad y atrofia del nervio óptico a los 3-6 meses de edad. Durante las primeras etapas, la tomografía computarizada (TC) revela una hiperdensidad característica en el tálamo y la corona radiata. Se cree que esto refleja células globoides de alta densidad y proliferación glial. La IRM también puede mostrar hiperintensidad en T1 e hipointensidad en T2 alrededor de los ventrículos, así como estructuras lineales similares a las que se observan en la LDM (Figura 3). El núcleo dentado cerebeloso, la sustancia blanca cerebelosa y el tracto piramidal del tronco cerebral muestran hiperintensidad T2 desde una fase temprana.
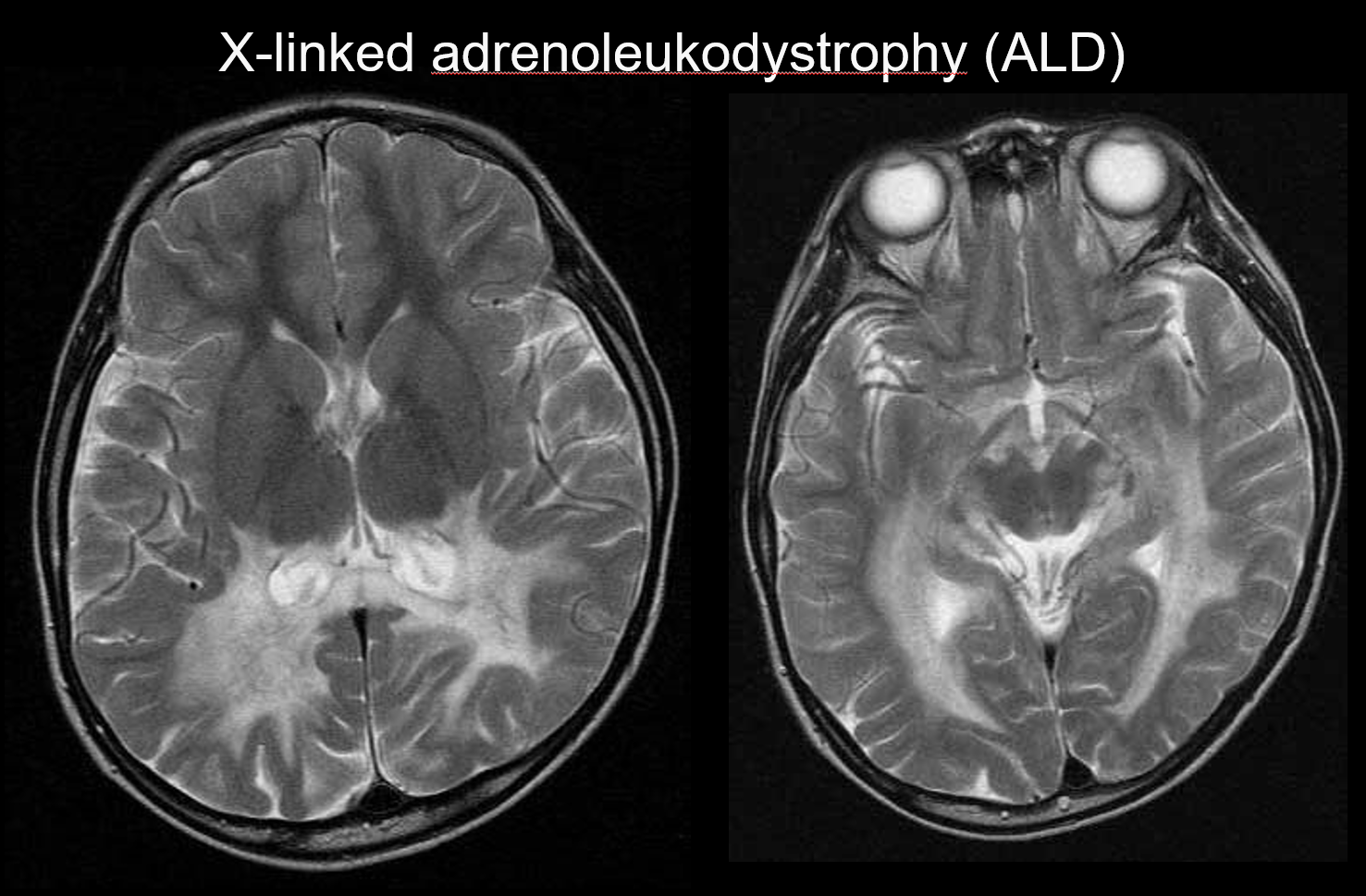
(b) Adrenoleucodistrofia ligada al cromosoma X
La adrenoleucodistrofia ligada al cromosoma X (ALD) es un trastorno hereditario recesivo ligado al cromosoma X (trastorno peroxisomal) causado por una anomalía del gen ABCD1 (cromosoma Xq28). La alteración de la β-oxidación provoca la acumulación de ácidos grasos de cadena muy larga en la sustancia blanca cerebral y en las glándulas suprarrenales, causando desmielinización e insuficiencia suprarrenal. La ALD se clasifica en las formas cerebral infantil, adolescente y adulta, la adrenomieloneuropatía (AMN) y la enfermedad de Addison únicamente. La forma cerebral infantil se desarrolla entre los 5 y los 8 años de edad, con la aparición de síntomas que incluyen el deterioro intelectual, la marcha espástica y el deterioro de la visión y la audición. Patológicamente, la desmielinización progresa desde la sustancia blanca que rodea el trígono del ventrículo lateral hasta el esplenio del cuerpo calloso, extendiéndose gradualmente de forma anterolateral. Como reflejo de la patología de la enfermedad, en la RM se aprecian hiperintensidades simétricas en T2 e hipointensidades en T1 que se extienden anterolateralmente desde la sustancia blanca que rodea el trígono del ventrículo lateral, con realce de contraste evidente en los márgenes (Figura 4). También son evidentes las lesiones del tracto corticoespinal.
4. Predominio periventricular
Estos trastornos se caracterizan principalmente por lesiones en la sustancia blanca que rodea los ventrículos laterales, estando preservada la sustancia blanca subcortical (fibras en U). Este patrón se observa en numerosos trastornos, incluida la LDM, y por lo tanto es comparativamente inespecífico. También se observan señales ligeramente anormales alrededor de los ventrículos laterales en la degeneración cortical, especialmente en las lipofuscinosis ceroides neuronales que se desarrollan después de la infancia.
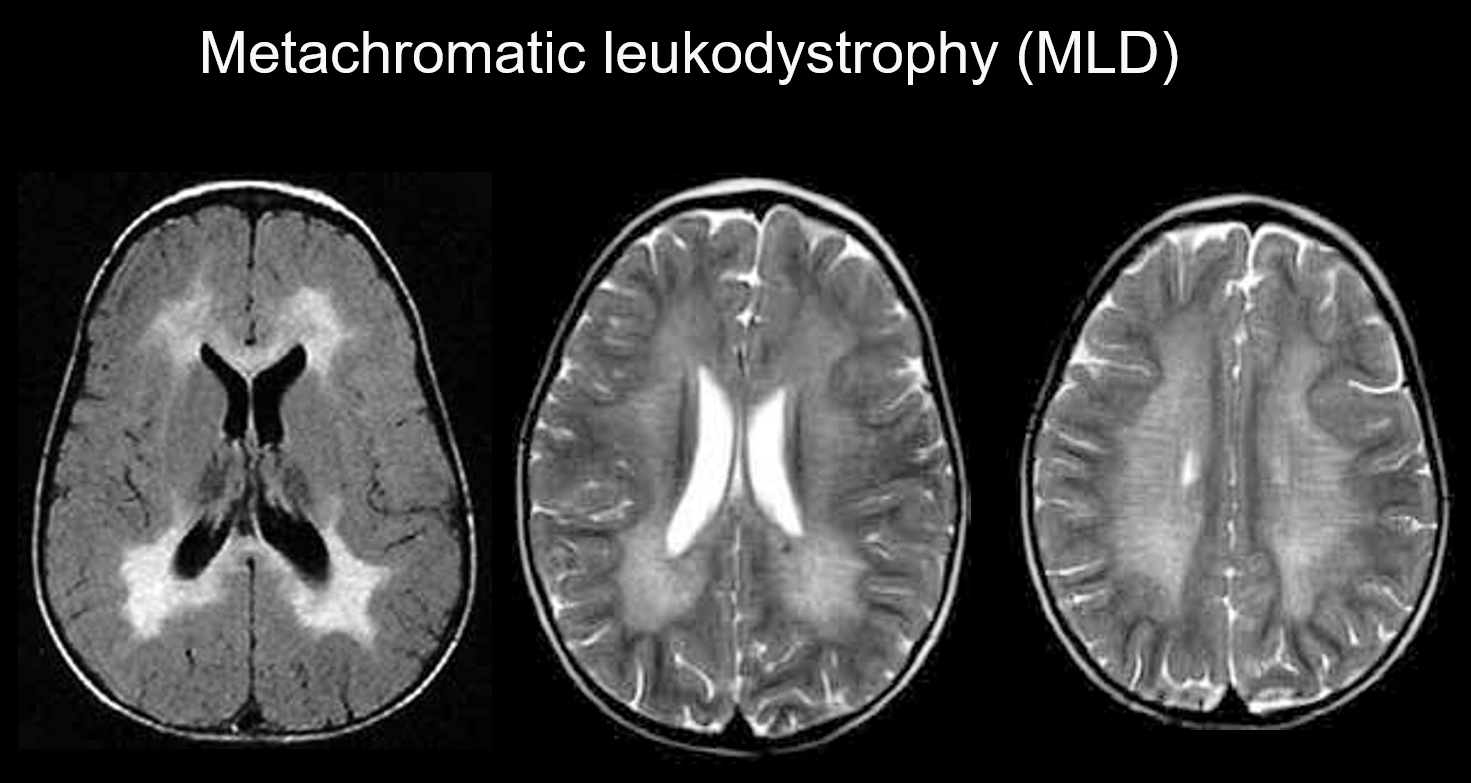
(a) Leucodistrofia metacromática.
La leucodistrofia metacromática es un trastorno hereditario autosómico recesivo (trastorno de almacenamiento lisosómico) causado por la deficiencia de arilsulfatasa-A (cromosoma 22q13.31), en el que la acumulación de sulfátidos altamente tóxicos provoca desmielinización. Dependiendo de la edad en la que aparece, se clasifica como congénita, de inicio infantil, de inicio juvenil o de inicio adulto. Sus síntomas incluyen regresión cognitiva, parálisis espástica, movimientos involuntarios, neuropatía periférica y atrofia del nervio óptico. Aparece en las imágenes ponderadas en T2 como hiperintensidades de la materia blanca, principalmente alrededor de los ventrículos laterales, y en las imágenes ponderadas en T1 como hipointensidades leves. Las lesiones tienden a ser predominantes en el lóbulo frontal. Dentro de las señales anormales generalizadas en la sustancia blanca pueden aparecer bandas de intensidad normal (rayas de tigre) (Figura 5). Se cree que se deben a la conservación parcial de la vaina de mielina en el espacio perivascular y a la acumulación de productos de degradación de la vaina de mielina en los macrófagos.
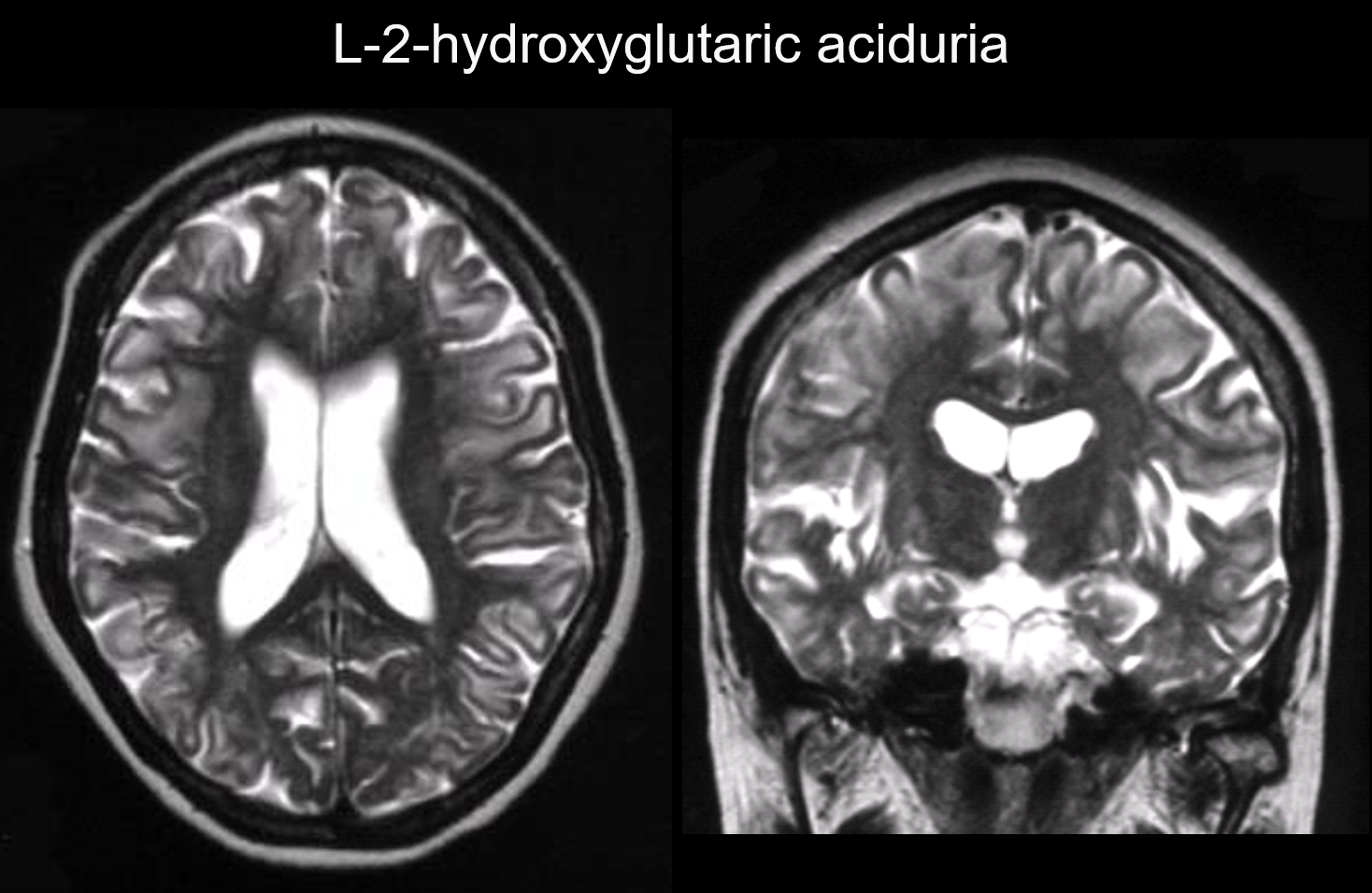
5. Predominio subcortical
En estos trastornos, las lesiones se producen principalmente en la sustancia blanca subcortical, incluidas las fibras en U. Los trastornos con este patrón incluyen la aciduria L-2-hidroxiglutárica (figura 6), la galactosemia, el síndrome de Kearns-Sayer, la academia propiónica, los trastornos del ciclo de la urea y la enfermedad de Canavan en fase inicial.
6. Cerebro difuso
En estos trastornos, aparecen señales anormales en toda la sustancia blanca cerebral. Presentan fuertes hiperintensidades T2 en comparación con las señales T2 producidas por la sustancia blanca no mielinizada (hipomielinización). Además de los casos de leucoencefalopatía megalencefálica con quistes subcorticales y leucoencefalopatía con desaparición de la sustancia blanca, los pacientes con cualquier tipo de trastorno de la sustancia blanca acaban mostrando este patrón a medida que avanza la enfermedad.
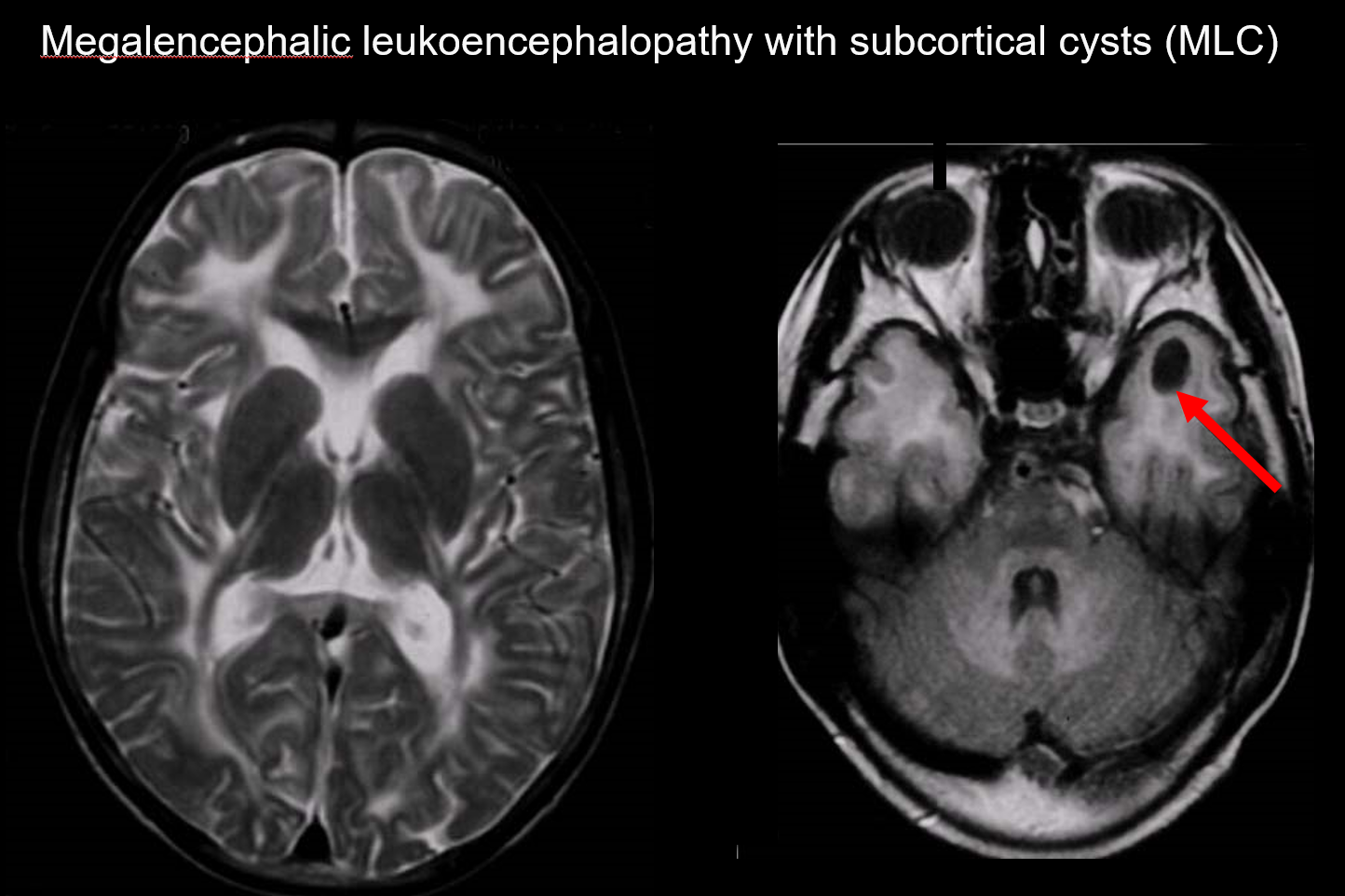
(a) Leucoencefalopatía megalencefálica con quistes subcorticales (MLC)
La MLC es un trastorno hereditario autosómico recesivo causado por una anomalía del gen MLC1, y su inicio en la infancia se caracteriza por megalocefalia, deterioro motor de progresión lenta, ataxia y espasticidad. La resonancia magnética revela señales anormales generalizadas características en la sustancia blanca y una leve inflamación de la misma, así como la formación de quistes en los lóbulos parietal y temporal (Figura 7).7, 8) Las imágenes ponderadas en T1 y T2 revelan una sustancia blanca anormal, mientras que todos los quistes muestran hipointensidad en T1 e hiperintensidad en T2, lo que los hace especialmente difíciles de detectar. Las imágenes FLAIR, que visualizan los quistes (agua) como hipointensidades, son valiosas para su diagnóstico. Es más común entre los japoneses que la leucoencefalopatía megalencefálica vacuolante.
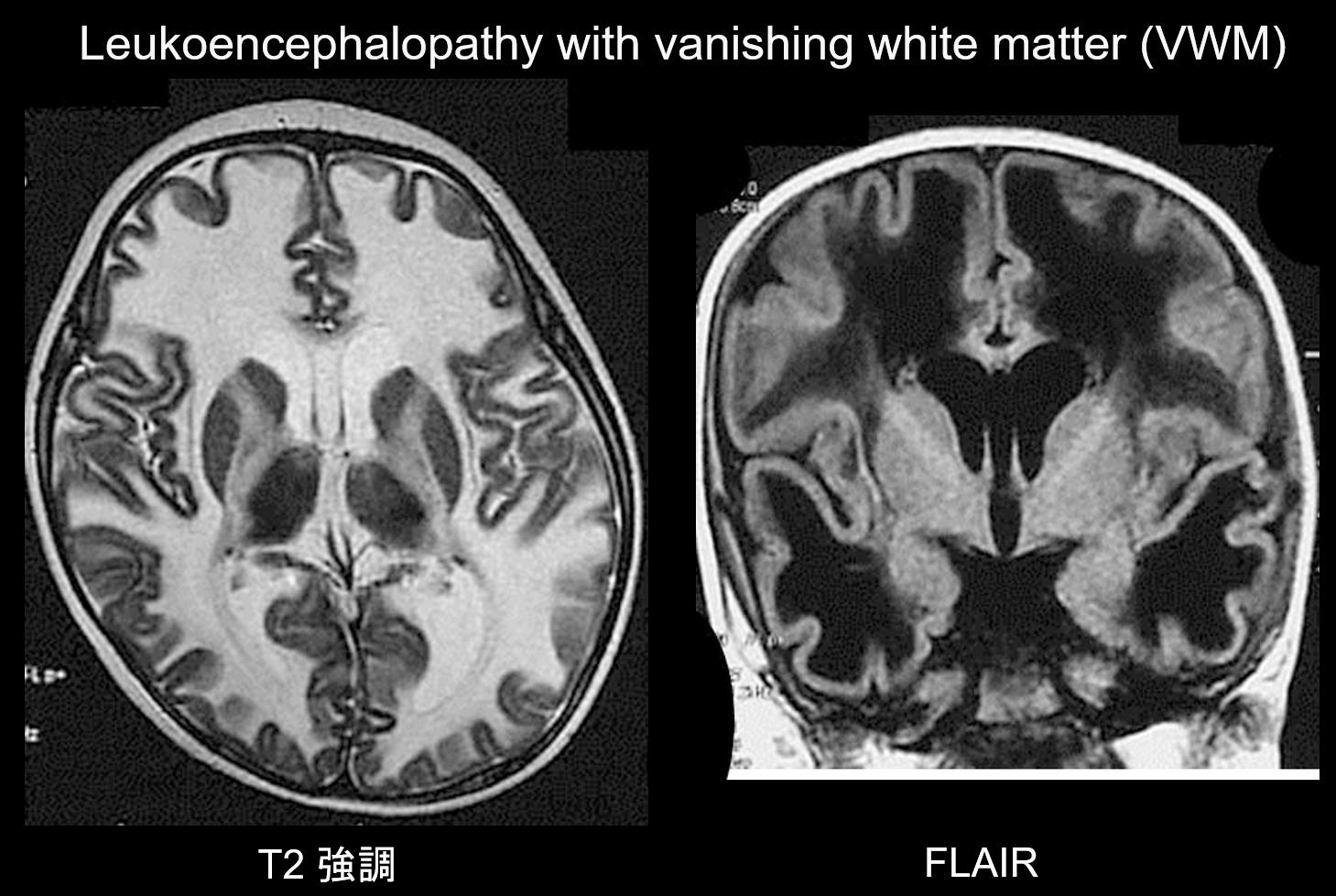
(b) Leucoencefalopatía con materia blanca evanescente.
La leucoencefalopatía con materia blanca evanescente (VWM) es un trastorno hereditario autosómico recesivo causado por una deficiencia de eIF2B, una proteína asociada a eIF2, que transfiere el ARNt iniciador a los ribosomas. eIF2B consta de cinco proteínas diferentes, todas ellas con loci genéticos distintos. Se ha demostrado que la VWM es el mismo trastorno que la ataxia cerebelosa infantil y la hipomielinización central (CACH). Los pacientes son normales durante el periodo neonatal y la primera infancia, pero después del inicio (normalmente a los 2-6 años) desarrollan una regresión cognitiva lentamente progresiva, espasticidad y ataxia. Se sabe que estos síntomas se agravan con las infecciones o los traumatismos menores. La sustancia blanca cerebral muestra una hiperintensidad T2 e hipointensidad T1 generalizadas y es sustituida gradualmente por líquido con el tiempo (como su nombre indica, la sustancia blanca desaparece) (Figura 8). La sustancia blanca quística contiene estructuras en banda que se cree que representan el tejido restante. También se observan señales anormales en el tronco encefálico, especialmente en el tracto tegmental central. Las imágenes FLAIR son valiosas para el diagnóstico de este trastorno.
7. Predominio o prominencia de la fosa posterior
Estos trastornos se caracterizan por lesiones predominantes en el tronco cerebral y el cerebelo. Las lesiones de la sustancia blanca cerebelosa pueden estar causadas por trastornos como la xantomatosis cerebrotendinosa (CTX), los trastornos peroxisomales, la enfermedad de Alexander, la leucoencefalopatía con afectación del tronco cerebral y la médula espinal y elevación del lactato (LBSL), la enfermedad de la orina con jarabe de arce, la histiocitosis y la toxicidad por heroína y cocaína. Las lesiones del tronco encefálico pueden estar causadas por trastornos como la enfermedad de Alexander, la LSBL y la poliglucosis del adulto. Las lesiones del pedúnculo cerebeloso medio se observan en el síndrome del cromosoma X frágil y en la leucodistrofia autosómica dominante del adulto relacionada con una duplicación de la lámina B1.
8. Lesiones multifocales
A diferencia de los trastornos que producen las lesiones confluentes descritas en el apartado 2-7 anterior, los trastornos de esta sección dan lugar a lesiones multifocales (dispersas). Incluyen infecciones como el síndrome TORCH (debido a una infección congénita por citomegalovirus u otra causa) y la brucelosis; trastornos inflamatorios como la encefalomielitis aguda diseminada (ADEM), la esclerosis múltiple (EM) y la neuromielitis óptica (NMO); vasculopatías como la arteriopatía cerebral autosómica dominante con infartos subcorticales y leucoencefalopatía (CADASIL), la aterosclerosis, la angiopatía amiloide, la enfermedad cerebral de pequeños vasos asociada a COL4A1, la enfermedad de Fabry y el síndrome de Susac y afecciones hereditarias como la enfermedad mitocondrial, la aciduria L-2-hidroxiglutárica, la mucopolisacaridosis (MPS) y las anomalías cromosómicas (como el síndrome 6p).
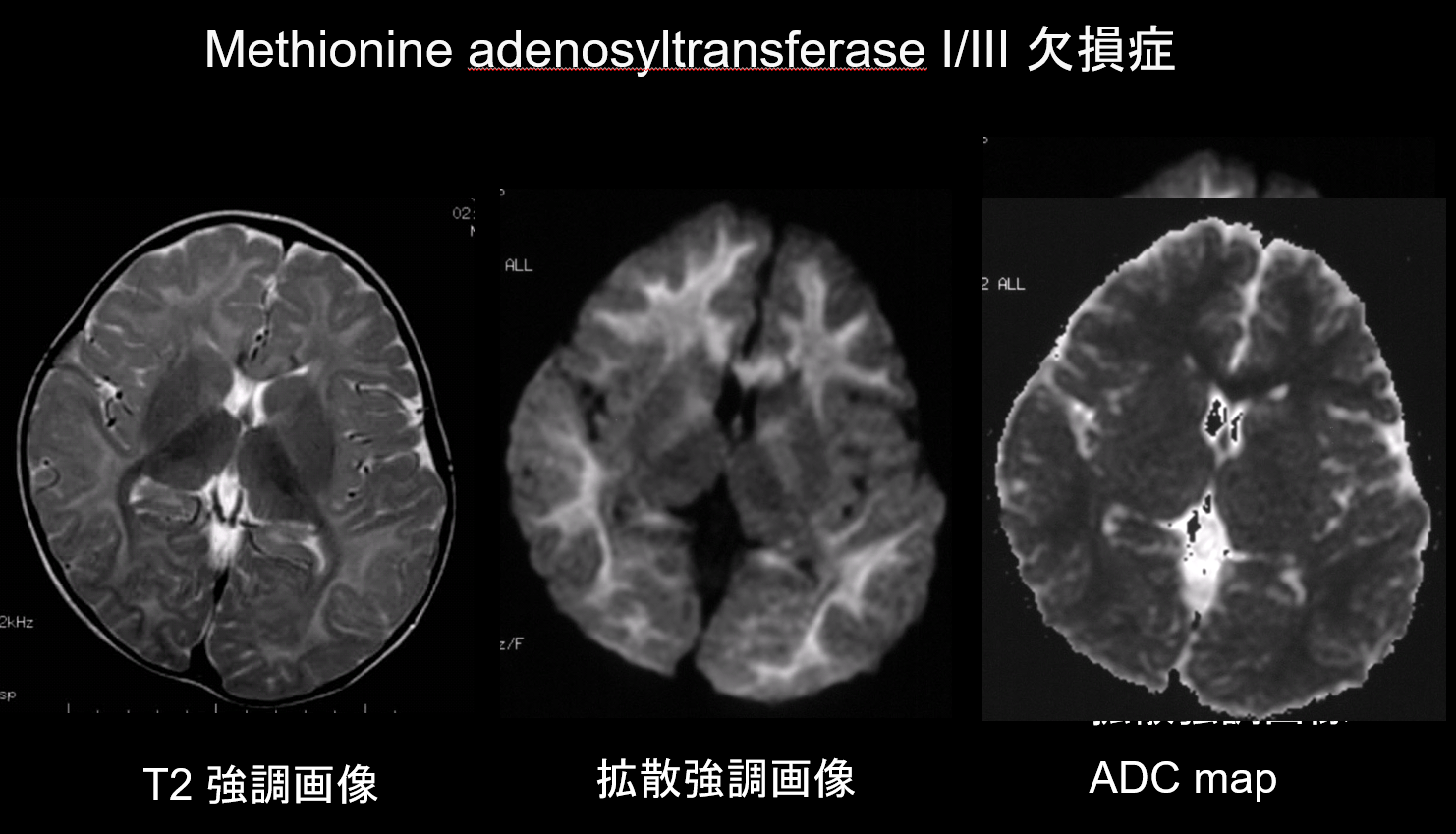
9. Lesiones con baja capacidad de difusión
Tanto en la desmielinización como en la hipomielinización, las principales patologías de los trastornos de la sustancia blanca, se produce una disminución de la cantidad de mielina, que limita la difusión, y el correspondiente aumento del líquido extracelular da lugar a hiperintensidades en T2 con un elevado coeficiente de difusión aparente (CDA). Es raro que los trastornos de la sustancia blanca presenten tanto hiperintensidades en T2 como un CDA bajo, por lo que esta combinación tiene un alto valor diagnóstico. Los trastornos caracterizados por la presencia de edema intramielínico dentro de la vaina de mielina y en los espacios entre vainas muestran un CDA bajo. Entre ellos se encuentran la enfermedad de la orina de jarabe de arce, la deficiencia de metionina adenosiltransferasa I/III (Figura 9), la fenilcetonuria, la hiperglicinemia no cetósica y la enfermedad de Canavan. La enfermedad de Krabbe y la leucodistrofia metacromática también pueden presentar un CDA bajo en algunas lesiones de la sustancia blanca, ya que puede producirse un edema intramielínico durante la fase aguda de la desmielinización.
- Van der Knaap MS, Valk J. Classification of myelin disorders. En Van der Knaap MS, Valk J, eds. Resonancia magnética de la mielinización y trastornos de la mielina. 3rd ed. Berlin: Springer, 2005, 20-24.
- Schiffmann R, van der Knaap MS. Un enfoque basado en la resonancia magnética para el diagnóstico de los trastornos de la materia blanca. Neurology 2009; 72: 750-759
- Takanashi J. Diagnostic imaging of white matter disorders. Revista de la Sociedad de Pediatría de Japón 2007; 111: 1243-1254.
- Van der Knaap MS, Breiter SN, Naidu S, et al. Definición y categorización de leucoencefalopatías de origen desconocido: MR imaging approach. Radiology 1999; 213: 121-133.
.